Tim and others, Regarding the depiction of protein cavities as per:
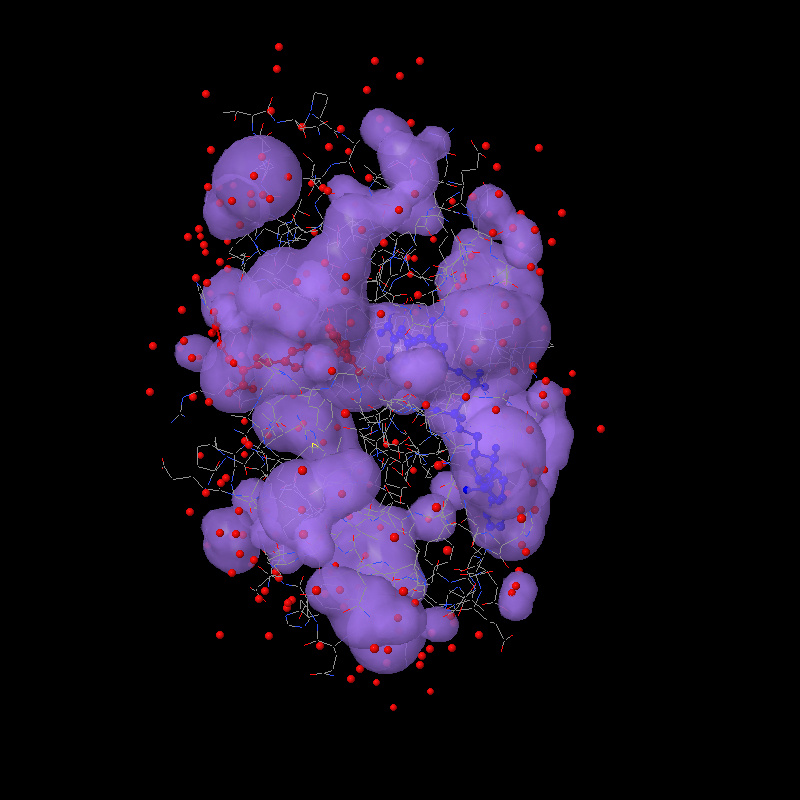
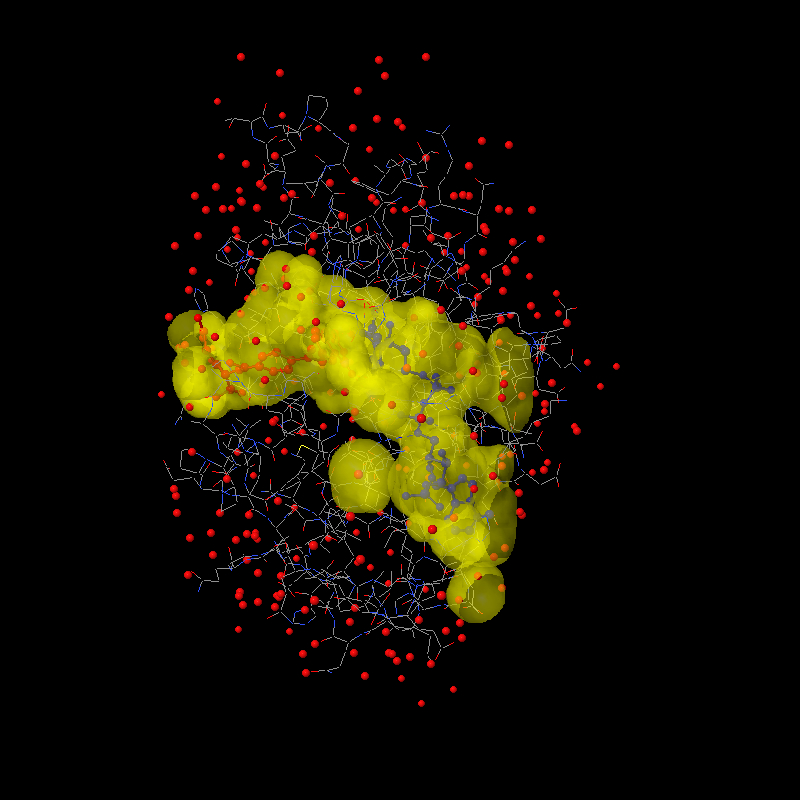
http://www.biochem.ucl.ac.uk/~roman/surfnet/examples/3dfr_site.html Good news! I tried a bunch of ideas this weekend, and then this morning I hit on the solution: The way solvent surfaces are generated in Jmol is probably unlike any other program. A grid of values is calculated just like for a molecular orbital, and then that grid is processed for the isosurface through the value 0. So values on the inside of the surface are < 0 and values on the outside are > 0. After quite some trial and error, I finally realized this morning that the key is those positive "outside" values. What we need to do to render cavities is (a) identify all grid points that have positive values AND are within the overall "envelope" of the protein and (b) use the value that is there for the RADIUS of a sphere at that point. That's your "foam filling"! Since each value measures the distance from the grid point to the nearest surface point, placing a sphere of that radius at that point perfectly "fits" the hole. It isn't important to fill a cavity with the perfect sphere -- the end goal is to turn around and use these grid-based spheres for ANOTHER solvent-surface calculation, this time as though we were trying to find the solvent surface around those "cavity" points. So then the other trick is how to identify just the cavity points and not all the other points far out from the molecule. The solution here uses the geosurface business that I set up. Actually, I sat down and cleaned up dots and geosurfaces so that their calculation could be used more generally. Here's how we identify the "envelope" of a protein: First we create a "dot surface" 10 angstroms out from all atoms. What you find is that only the outer atoms contribute to this -- all the other atoms are hidden. Then the distance from each "foam cavity sphere" to the nearest surface dot is measured. If this distance is < 10, then we can throw out the point, because the point must be "outside" the molecule. But if the distance is > 10, then that point must be "within" the molecule. We keep it. What do you know? Perfect rendering of cavities as if they themselves were atoms. Or, at least I hope you think so. Here's a jpg to get your appetite whetted: http://www.stolaf.edu/academics/chemapps/jmol/docs/misc/3dfr-a.jpg http://www.stolaf.edu/academics/chemapps/jmol/docs/misc/3dfr-b.jpg The first was done using: load =1dfr;select protein;spacefill off;wireframe on;color (ndp) blue;color (mtx) red isosurface select(protein) ignore(not protein) cavity 10 1.4 color isosurface translucent 4 write image 800 800 1dfr-a.jpg The second focuses on the two ligands and uses: isosurface isosurface1 select(protein and within(4.0, not protein)) ignore(not protein) cavity 10 1.2 color isosurface translucent 6 yellow write image 800 800 1dfr-b.jpg The two parameters are the envelope radius (10 effectively defines the radius of the outer "pores" that define a cavity) and the minimum cavity radius (1.2 Angstroms about the size of a water molecule). These parameters can be tweaked. Maybe Tim, you can help me settle on some reasonable defaults. Bob ------------------------------------------------------------------------- Take Surveys. Earn Cash. Influence the Future of IT Join SourceForge.net's Techsay panel and you'll get the chance to share your opinions on IT & business topics through brief surveys-and earn cash http://www.techsay.com/default.php?page=join.php&p=sourceforge&CID=DEVDEV _______________________________________________ Jmol-developers mailing list [email protected] https://lists.sourceforge.net/lists/listinfo/jmol-developers
{kind=link}
{kind=link}