questions answered in line below...
On Thu, Nov 11, 2010 at 7:57 AM, FyD <f...@q4md-forcefieldtools.org> wrote:
> Dear Bob, dear Angel,
>
>
> As you can see
> http://cluster.q4md-forcefieldtools.org/~ucpublic1/javaappletpdb-1.html<http://cluster.q4md-forcefieldtools.org/%7Eucpublic1/javaappletpdb-1.html>
> is ok, while
> http://cluster.q4md-forcefieldtools.org/~ucpublic1/javaappletpdb-2.html<http://cluster.q4md-forcefieldtools.org/%7Eucpublic1/javaappletpdb-2.html>
> leads to ugly connections; I guess this because of the absence of
> chemical elements in Mol_m2-o1-qmra.pdb.
>
> Why does Jmol need chemical elements to recognize atom names?
>
Jmol doesn't need atom symbols, but as you can imagine, CA for calcium and
CA for alpha carbon is not conducive to unambiguous resolution of elements
without that key field. There are no rules about atom names having to start
with element symbols, so Jmol can only apply a reasonable guess algorithm to
get elements when that element field data is missing.
- Many small molecules are written in the PDB file format without
> these chemical elements
>
Yes, but for them the atom name "CA1" is interpreted as calcium, as you
would expect. Same for Ce1, Cd1, Cf1. How could Jmol be expected to read
those as carbons?
> - VMD and LEaP (for instance) do not need chemical elements to
> recognize atom names
>
>
And they will make the same mistake -- or not recognize cerium as a possible
atom. VMD has all sorts of known problems figuring out elements from atom
names.
> Do you think it would be possible that Jmol recognizes atom names
> without chemical elements?
>
>
Jmol does this already. but you can't throw generic "PDB-like" files at it
and expect it to know that CA1 means alpha carbon and not calcium. One could
suggest that ATOM requires CA be alpha carbon and HETATM might not, but it
turns out that's not workable, either.
Jmol recognizes that if you start "CA1" in column 13, then you must mean
calcium, and if you start it in column 14, you mean alpha carbon. That is,
columns 12 and 13 indicate atom symbol. So that's an option. (The atom names
are spit out by many programs in this way -- all the RCSB PDB files are like
this, for example, even if it is not in the specs.) This system breaks down
for certain hydrogen atoms that are named as, for example, "HH11" or "HO5'".
For "HO5'" Jmol checks to see if this is a HETATM (so this is read as
holmium, or ATOM (so this is read as hydrogen). Obviously the best situation
is to create proper PDB files and have that colum 77/78 data.
---
>
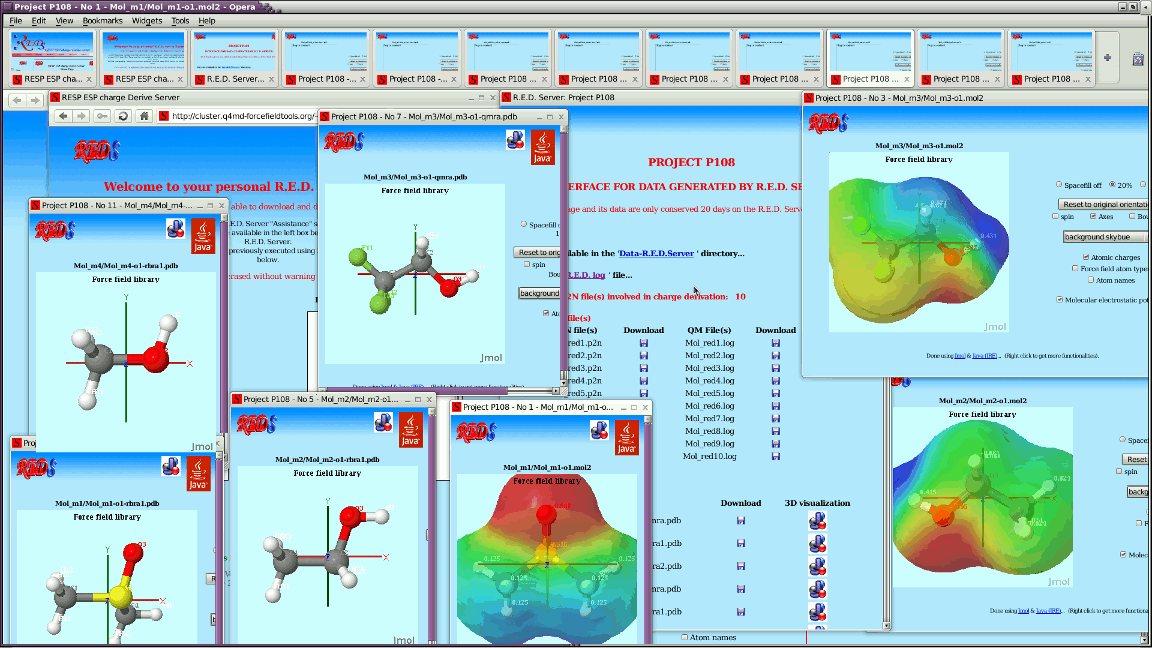
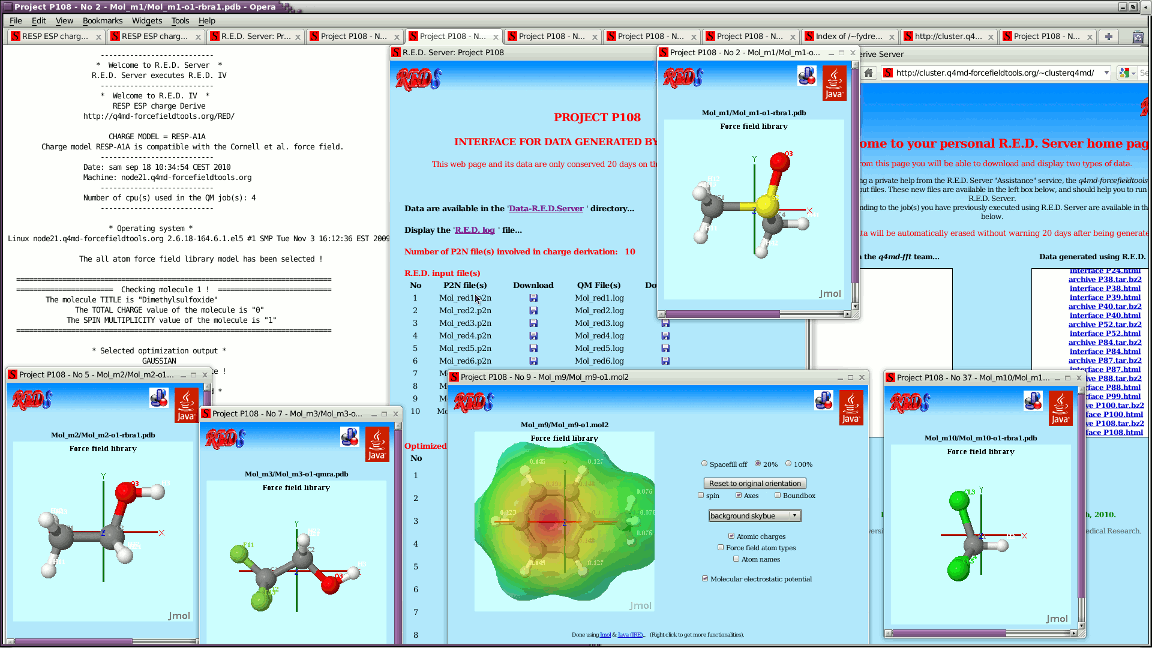
> For each job, R.E.D. Server generates by now a Jmol-based graphical
> interface to display outputs generated by R.E.D. IV.
> See for instance:
> http://q4md-forcefieldtools.org/Tutorial/DNA-FFTopDB/P1.html
> &
> http://q4md-forcefieldtools.org/REDS/images/Screenshot1.gif
> http://q4md-forcefieldtools.org/REDS/images/Screenshot2.gif
> http://q4md-forcefieldtools.org/REDS/images/Screenshot3.gif
>
> The problem is that some of the applets generated for the PDB files
> present broken/bad atom connectivities because of the absence of
> chemical elements; we can obviously add chemical elements in our PDB
> files (no problem) - however doing so in the P2N file format might not
> be that straightforward...
>
>
Why is that?
> I continue now with these simple atom names
> ATOM 1 C1 BNZ 1 0.5624 1.2144 0.3611
> ATOM 2 H1 BNZ 1 0.9988 2.1568 0.6413
> ATOM 3 C2 BNZ 1 -0.7794 1.1461 0.0200
> ATOM 4 H2 BNZ 1 -1.3843 2.0354 0.0356
> ATOM 5 C3 BNZ 1 -1.3418 -0.0683 -0.3411
> ATOM 6 H3 BNZ 1 -2.3830 -0.1213 -0.6057
> ATOM 7 C4 BNZ 1 -0.5624 -1.2144 -0.3611
> ATOM 8 H4 BNZ 1 -0.9988 -2.1568 -0.6413
> ATOM 9 C5 BNZ 1 0.7794 -1.1461 -0.0200
> ATOM 10 H5 BNZ 1 1.3843 -2.0354 -0.0356
> ATOM 11 C6 BNZ 1 1.3418 0.0683 0.3410
> ATOM 12 H6 BNZ 1 2.3830 0.1213 0.6057
> no problem: atom connectivites are correct in a Jmol applet even in
> the absence of chemical elements; it also works for
>
> http://q4md-forcefieldtools.org/Tutorial/DNA-FFTopDB/P1/javaappletpdb-6.html
> where atom names are not that simple.
>
> Consequently, this means that Jmol can already recognize some atom
> names without chemical elements. Is it not possible to extend this
> feature to more general atom names?
>
Right, that's because Jmol has a fallback option. The rule is that the atom
symbol should appear right-justified in
columns 13 and 14. But if that's not the case, as here, then the algorithm
looks next at just the character in
column 13. In this case C and H.
By the way, are you sure ATOM is appropriate for a benzene ring? According
to the specs we have:
The ATOM records present the atomic coordinates for standard residues (see
http://deposit.pdb.org/public-component-erf.cif).
Everything else should be HETATM. The only difference that makes for Jmol is
for four-letter
atom names that are therefore forced to start in column 13, start with H,
but aren't hydrogen. Like,
for example, HE for helium or "hydrogen E". In that case, if it is HETATM,
Jmol chooses helium; if
it is ATOM, it chooses H.
Bob
>
>
> ------------------------------------------------------------------------------
> Centralized Desktop Delivery: Dell and VMware Reference Architecture
> Simplifying enterprise desktop deployment and management using
> Dell EqualLogic storage and VMware View: A highly scalable, end-to-end
> client virtualization framework. Read more!
> http://p.sf.net/sfu/dell-eql-dev2dev
> _______________________________________________
> Jmol-users mailing list
> Jmol-users@lists.sourceforge.net
> https://lists.sourceforge.net/lists/listinfo/jmol-users
>
--
Robert M. Hanson
Professor of Chemistry
St. Olaf College
1520 St. Olaf Ave.
Northfield, MN 55057
http://www.stolaf.edu/people/hansonr
phone: 507-786-3107
If nature does not answer first what we want,
it is better to take what answer we get.
-- Josiah Willard Gibbs, Lecture XXX, Monday, February 5, 1900
------------------------------------------------------------------------------
Centralized Desktop Delivery: Dell and VMware Reference Architecture
Simplifying enterprise desktop deployment and management using
Dell EqualLogic storage and VMware View: A highly scalable, end-to-end
client virtualization framework. Read more!
http://p.sf.net/sfu/dell-eql-dev2dev
_______________________________________________
Jmol-users mailing list
Jmol-users@lists.sourceforge.net
https://lists.sourceforge.net/lists/listinfo/jmol-users
{kind=link}
{kind=link}
{kind=link}