Alan wrote :
>Already in the original Rietveld program, the 'observed' Bragg intensities
(or
>structure factors) were estimated by dividing the intensity at each profile
>point according to the calculated structure factors. This is more useful
than
>it might seem :-) since by iteration of difference Fouriers, such ESTIMATED
>'observed' Bragg intensities can be used to obtain a more complete model for
>Rietveld refinement eg to locate hydrogen atoms in neutron diffraction. (The
>original purpose was just to estimate the conventional Bragg R-factor).
Just a comment. If the "normal" method is the classic Rietveld method, this
cannot be considered as a method for extracting structure factors. This is a
method for extracting structure factors knowing already a large part of the
structure model, not exactly the same.
>A second method was invented by Pawley while Armel was still a young man :-)
>Pawley, G. S. (1980) J. Appl. Crystallogr. 13, 630-3.
>"EDINP, the Edinburgh powder profile refinement program ."
In the above sense, the Pawley method is not a second method. It is the first
method allowing to extract structure factors with cell constraint, without any
prior structure knowledge. Nevertheless, methods for extracting structure
factors without cell constraint were existing much before the Pawley and
Rietveld methods.
The problem was that it could take a long time to extract 1000 structure
factors, almost one by one ! Moreover, the lack of cell constraint led to
(more) inaccuracy when dealing with strongly overlaping reflections.
>In this Pawley method, the individual Bragg intensities are refined to fit
the
>full diffraction pattern, a kind of "profile refinement" that does not
require
a
>model for the structure, and that produces a less biased estimate than does
>the original Rietveld method ("normal method").
Nobody can use the "normal method" when the structure is unknown. So
that the "normal method" simply does not exist.
>The Pawley method was then improved by le Bail, and it is now Armel's code
>that is commonly used.
I never improved the Pawley method which is continuing its own route. My
method
consists in iterating the Rietveld decomposition formula, starting from |F|
values
arbitrarily set to be all identical. So that, this is the second cell
constrained method
for extracting structure factors without any prior structure model knowledge.
It
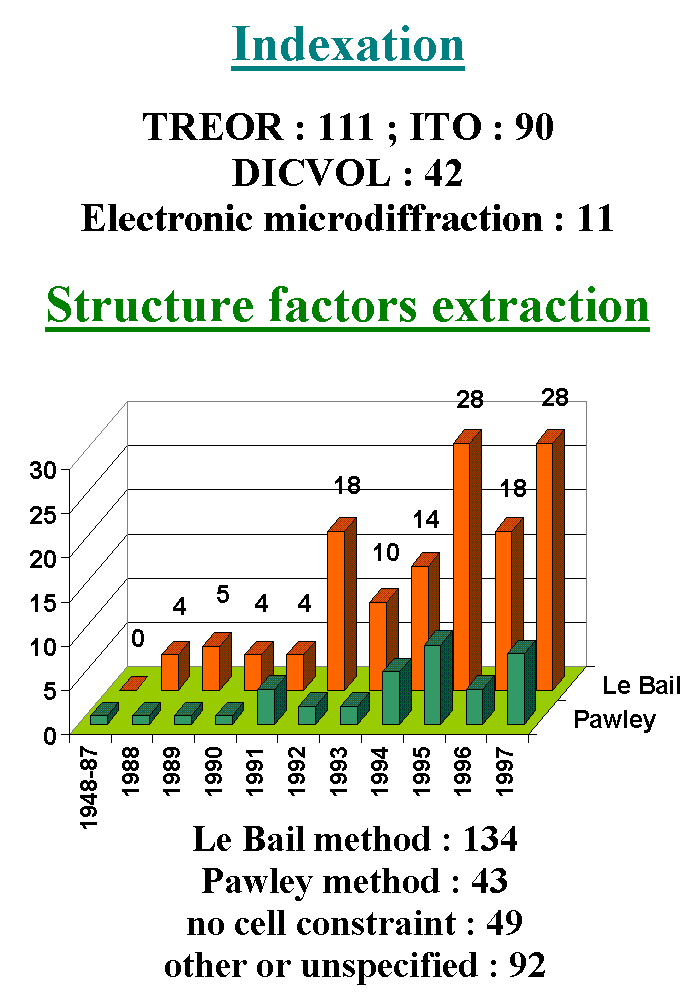
is true that my method is statistically more used than the Pawley method (the
ratio is exactly 134/43 up to 1997 included :)
http://www.cristal.org/iniref/ecm18/t6.gif
As Bill David would say, people are using available programs. Programs
having implemented (and improved) the Pawley method are not numerous,
nor easily available... Many Rietveld codes have included the Le Bail
method as an option (FULLPROF, GSAS, MPROF, ARIT, etc) and
standalone programs exist as well (EXTRA, EXTRACT...), explaining its
more common use.
Last point, but not least, the Pawley method, although published in
1980, was applied for the first time to an ab initio structure determination
by powder diffractometry in 1987 (and, yes, I was young : 37 ;-). Compared
to Alan, I will stay young, I guess.
Armel Le Bail - Universit� du Maine, Laboratoire des Fluorures,
CNRS ESA 6010, Av. O. Messiaen, 72085 Le Mans Cedex 9, France
http://www.cristal.org/
{kind=link}