On 04/25/2011 07:04 AM, O. Baris Malcioglu wrote: > Dear Hongsheng Zhao, > > Unless you are asking if someone ever tried forcing a particular > "bond" or aromaticity in PW explicitly (one can try to constrain some > number of KS states trying to estimate a particular bond, but I don't > think this is a good idea to do in a pw DFT code), I think you might > be a little confused by the concepts. > > Beware that in literature they tend to do assume hydrogens implicitly > in order not to complicate figure presentation. Also, please notice > that you add hydrogens not just to saturate, but to put the atom in a > particular "bonding state" (i.e. sp3 or sp2 hybridization etc.). That > "bonding state" is handled by PW with no additional constraints (i.e. > when you input the coordinates of Carbon and Hydrogen atoms in > Benzene, with a suitable XC kernel, the electronic structure will > assume aromaticity without you explicitly mentioning it, and if you > want to see explicit proof of aromaticity using the nomenclature of > theoretical chemistry, you can, for example, project the ks states in > terms of atom centered pi levels) Thanks a lot for your kindly reply. In fact, I want to know how to do the corresponding adding operations in the input file for pwscf.
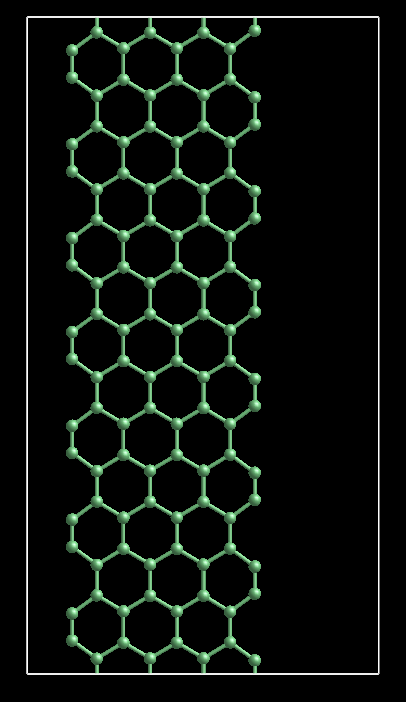
Say, for example, the following is the model input file for graphene nanoribbon including 7 primitive unit cell of armchair graphene nanoribbon (8), which is described and used in the following paper: http://tinyurl.com/3nz3xz9 , --------------- &CONTROL calculation = 'scf' prefix='block_1' pseudo_dir = './' outdir='./' tstress=.true. tprnfor = .true. forc_conv_thr=1.0d-4 nstep=200 / &SYSTEM ibrav=0 celldm(1)=1.889726 nat=112 ntyp=1 ecutwfc=36 / &ELECTRONS mixing_beta=0.7 conv_thr = 1.0d-8 electron_maxstep=200 / &IONS trust_radius_max=0.2 / &CELL cell_dynamics='bfgs' / ATOMIC_SPECIES C 12.0110 C.UPF CELL_PARAMETERS (alat) 16.5000000000 0.0000000000 0.0000000000 0.0000000000 8.0000000000 0.0000000000 0.0000000000 0.0000000000 30.8203000000 ATOMIC_POSITIONS (crystal) C 0.128008000 0.500000000 0.050828000 C 0.128008000 0.500000000 0.092030000 C 0.197928000 0.500000000 0.023239000 C 0.197929000 0.500000000 0.119619000 C 0.273757000 0.500000000 0.047731000 C 0.273757000 0.500000000 0.095127000 C 0.349589000 0.500000000 0.023674000 C 0.349589000 0.500000000 0.119183000 C 0.425569000 0.500000000 0.047753000 C 0.425570000 0.500000000 0.095104000 C 0.501414000 0.500000000 0.023700000 C 0.501414000 0.500000000 0.119157000 C 0.577255000 0.500000000 0.048189000 C 0.577255000 0.500000000 0.094668000 C 0.647174000 0.500000000 0.020600000 C 0.647174000 0.500000000 0.122256000 C 0.128008000 0.500000000 0.193686000 C 0.128008000 0.500000000 0.234887000 C 0.197928000 0.500000000 0.166096000 C 0.197929000 0.500000000 0.262476000 C 0.273757000 0.500000000 0.190588000 C 0.273757000 0.500000000 0.237984000 C 0.349589000 0.500000000 0.166531000 C 0.349589000 0.500000000 0.262040000 C 0.425569000 0.500000000 0.190611000 C 0.425570000 0.500000000 0.237961000 C 0.501414000 0.500000000 0.166557000 C 0.501414000 0.500000000 0.262014000 C 0.577255000 0.500000000 0.191046000 C 0.577255000 0.500000000 0.237525000 C 0.647174000 0.500000000 0.163457000 C 0.647174000 0.500000000 0.265113000 C 0.128008000 0.500000000 0.336543000 C 0.128008000 0.500000000 0.377744000 C 0.197928000 0.500000000 0.308953000 C 0.197929000 0.500000000 0.405334000 C 0.273757000 0.500000000 0.333445000 C 0.273757000 0.500000000 0.380842000 C 0.349589000 0.500000000 0.309389000 C 0.349589000 0.500000000 0.404897000 C 0.425569000 0.500000000 0.333468000 C 0.425570000 0.500000000 0.380818000 C 0.501414000 0.500000000 0.309414000 C 0.501414000 0.500000000 0.404871000 C 0.577255000 0.500000000 0.333903000 C 0.577255000 0.500000000 0.380382000 C 0.647174000 0.500000000 0.306315000 C 0.647174000 0.500000000 0.407970000 C 0.128008000 0.500000000 0.479400000 C 0.128008000 0.500000000 0.520601000 C 0.197928000 0.500000000 0.451810000 C 0.197929000 0.500000000 0.548191000 C 0.273757000 0.500000000 0.476302000 C 0.273757000 0.500000000 0.523699000 C 0.349589000 0.500000000 0.452246000 C 0.349589000 0.500000000 0.547755000 C 0.425569000 0.500000000 0.476325000 C 0.425570000 0.500000000 0.523675000 C 0.501414000 0.500000000 0.452271000 C 0.501414000 0.500000000 0.547729000 C 0.577255000 0.500000000 0.476760000 C 0.577255000 0.500000000 0.523239000 C 0.647174000 0.500000000 0.449172000 C 0.647174000 0.500000000 0.550827000 C 0.128008000 0.500000000 0.622257000 C 0.128008000 0.500000000 0.663458000 C 0.197928000 0.500000000 0.594667000 C 0.197929000 0.500000000 0.691048000 C 0.273757000 0.500000000 0.619159000 C 0.273757000 0.500000000 0.666556000 C 0.349589000 0.500000000 0.595103000 C 0.349589000 0.500000000 0.690612000 C 0.425569000 0.500000000 0.619182000 C 0.425570000 0.500000000 0.666532000 C 0.501414000 0.500000000 0.595128000 C 0.501414000 0.500000000 0.690586000 C 0.577255000 0.500000000 0.619617000 C 0.577255000 0.500000000 0.666096000 C 0.647174000 0.500000000 0.592029000 C 0.647174000 0.500000000 0.693684000 C 0.128008000 0.500000000 0.765114000 C 0.128008000 0.500000000 0.806316000 C 0.197928000 0.500000000 0.737525000 C 0.197929000 0.500000000 0.833905000 C 0.273757000 0.500000000 0.762016000 C 0.273757000 0.500000000 0.809413000 C 0.349589000 0.500000000 0.737960000 C 0.349589000 0.500000000 0.833469000 C 0.425569000 0.500000000 0.762039000 C 0.425570000 0.500000000 0.809389000 C 0.501414000 0.500000000 0.737985000 C 0.501414000 0.500000000 0.833443000 C 0.577255000 0.500000000 0.762475000 C 0.577255000 0.500000000 0.808953000 C 0.647174000 0.500000000 0.734886000 C 0.647174000 0.500000000 0.836542000 C 0.128008000 0.500000000 0.907971000 C 0.128008000 0.500000000 0.949173000 C 0.197928000 0.500000000 0.880382000 C 0.197929000 0.500000000 0.976762000 C 0.273757000 0.500000000 0.904873000 C 0.273757000 0.500000000 0.952270000 C 0.349589000 0.500000000 0.880817000 C 0.349589000 0.500000000 0.976326000 C 0.425569000 0.500000000 0.904896000 C 0.425570000 0.500000000 0.952246000 C 0.501414000 0.500000000 0.880842000 C 0.501414000 0.500000000 0.976300000 C 0.577255000 0.500000000 0.905332000 C 0.577255000 0.500000000 0.951810000 C 0.647174000 0.500000000 0.877743000 C 0.647174000 0.500000000 0.979399000 K_POINTS {automatic} 4 4 4 1 1 1 --------------- In that paper, we can find the following description on page two: --------- The carbon atoms at the edges are saturated with H atoms. ---------- So, according to the above description, what should I revise the input file in order to saturating the edges with H atoms? Attached please find the snapshot for this structure. Regards. -- Hongsheng Zhao<zhaohscas at yahoo.com.cn> School of Physics and Electrical Information Science, Ningxia University, Yinchuan 750021, China -------------- next part -------------- A non-text attachment was scrubbed... Name: agnr8.png Type: image/png Size: 65504 bytes Desc: not available Url : http://www.democritos.it/pipermail/pw_forum/attachments/20110426/d61023dd/attachment-0001.png
{kind=link}