Sir i m doing on theoretical research work . plz suggest me how to find DFT calculation and band structure from quantum espresso
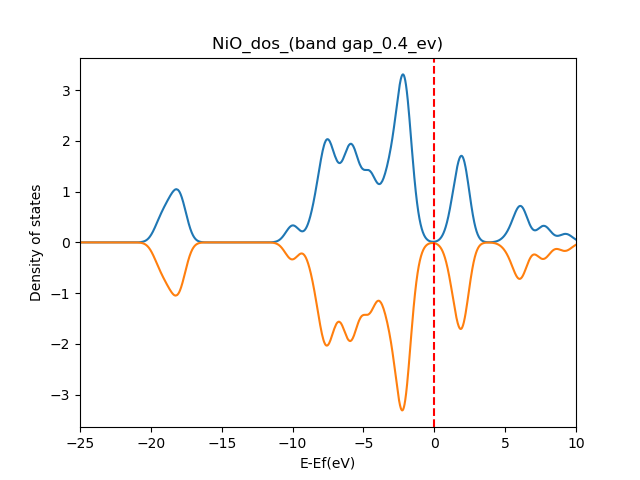
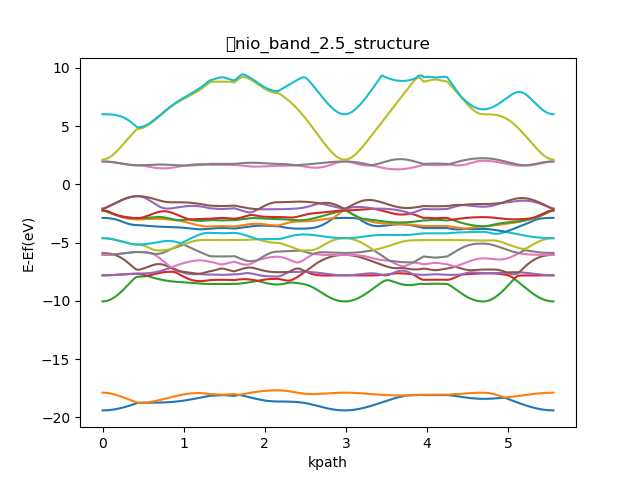
On Wed, Jan 22, 2020 at 4:32 PM <[email protected]> wrote: > Send users mailing list submissions to > [email protected] > > To subscribe or unsubscribe via the World Wide Web, visit > https://lists.quantum-espresso.org/mailman/listinfo/users > or, via email, send a message with subject or body 'help' to > [email protected] > > You can reach the person managing the list at > [email protected] > > When replying, please edit your Subject line so it is more specific > than "Re: Contents of users digest..." > > > Today's Topics: > > 1. Re: QE 6.4 Error signal SIGSEGV: Segmentation fault invalid > memory reference - Pt12-Rh cluster relax calculation (Paolo > Giannozzi) > 2. mod value in polarization calculation with Berry phase > ([email protected]) > 3. NiO band structure and density of states (Poonam Kaushik) > 4. Re: NiO band structure and density of states (Timrov Iurii) > > > ---------------------------------------------------------------------- > > Message: 1 > Date: Tue, 21 Jan 2020 14:30:30 +0100 > From: Paolo Giannozzi <[email protected]> > To: barunachalam <[email protected]>, Quantum ESPRESSO users Forum > <[email protected]> > Subject: Re: [QE-users] QE 6.4 Error signal SIGSEGV: Segmentation > fault invalid memory reference - Pt12-Rh cluster relax calculation > Message-ID: > <CAPMgbCsAReYy= > [email protected]> > Content-Type: text/plain; charset="utf-8" > > It seems to be a problem that occurs when the data file is written at the > end of a non-converged run. I vaguely remember that something similar was > corrected some time ago. You should try a more recent version. > > Paolo > > > On Tue, Jan 21, 2020 at 7:20 AM barunachalam <[email protected]> wrote: > > > Dear Sir, > > > > I am getting the error *Program received signal SIGSEGV: Segmentation > > fault - invalid memory reference, *during relax calculation of Pt12-Rh > > structure. However, in case Pt12-Au structure, there is no error and it > is > > converging fully. > > > > I would like to know whether, the issue with &control, &electron, &ion > > configuration or any software issue. > > > > Thanks and Regards, > > > > B.Arunachalam > > > > Research Scholar, > > > > NIT Trichy > > > > > > > > *Configuration sample* > > > > &system > > > > ibrav = 1, celldm(1) =30.0, nat= 13, ntyp= 2, > > > > ecutwfc = 30.0,ecutrho = 300.0, > > > > report=1, > > > > occupations='smearing', smearing='marzari-vanderbilt', degauss=0.05, > > > > noncolin = .false., > > > > nspin=2, > > > > nbnd=69, > > > > starting_magnetization(1) = 0.5 > > > > starting_magnetization(2) = 0.5 > > > > / > > > > &electrons > > > > mixing_beta = 0.7 > > > > conv_thr = 1.0d-08 > > > > electron_damping=0.001, > > > > ortho_max=100, > > > > emass=500, > > > > emass_cutoff=3., > > > > electron_velocities='zero', > > > > / > > > > &ions > > > > ion_dynamics='damp', > > > > ion_velocities='zero', > > > > ion_radius(1)=1.0, > > > > ion_damping=0.01 > > > > *Error information* > > > > total energy = -9255.91088445 Ry > > > > Harris-Foulkes estimate = -9255.91088451 Ry > > > > estimated scf accuracy < 0.00000002 Ry > > > > total magnetization = 0.02 Bohr mag/cell > > > > absolute magnetization = 0.15 Bohr mag/cell > > > > End of self-consistent calculation > > > > convergence NOT achieved after 100 iterations: stopping > > > > Writing output data file PtRu_pbe.save/ > > > > *Program received signal SIGSEGV: Segmentation fault - invalid memory > > reference.* > > > > Backtrace for this error: > > > > #0 0x7F34E6A1E697 > > > > #1 0x7F34E6A1ECDE > > > > #2 0x7F34E5F1A2EF > > > > #3 0xA2324C in __fox_m_fsys_format_MOD_real_dp_str at > > fox_m_fsys_format.F90:? > > > > #4 0xA22029 in __fox_m_fsys_format_MOD_str_real_dp_fmt at > > fox_m_fsys_format.F90:? > > > > #5 0xA20F05 in __fox_m_fsys_format_MOD_str_real_dp_array_fmt at > > fox_m_fsys_format.F90:? > > > > #6 0xA20BA5 in __fox_m_fsys_format_MOD_str_real_dp_array_fmt_chk > > > > #7 0x9D909F in __m_wxml_overloads_MOD_charactersarrayrealdp > > > > #8 0x74661D in __qes_write_module_MOD_qes_write_matrix.part.2 at > > qes_write_module.f90:2462 > > > > #9 0x75112A in qes_write_matrix at qes_write_module.f90:320 > > > > #10 0x4F2679 in __pw_restart_new_MOD_pw_write_schema at > > pw_restart_new.f90:659 > > > > #11 0x4E5BAE in punch_ at punch.f90:66 (discriminator 1) > > > > #12 0x50B1F9 in run_pwscf_ at run_pwscf.f90:142 > > > > #13 0x409243 in MAIN__ at pwscf.f90:98 > > > > > > > =================================================================================== > > > > = BAD TERMINATION OF ONE OF YOUR APPLICATION PROCESSES > > > > = PID 417219 RUNNING AT ssl_hn > > > > = EXIT CODE: 139 > > > > = CLEANING UP REMAINING PROCESSES > > > > = YOU CAN IGNORE THE BELOW CLEANUP MESSAGES > > > > > > > =================================================================================== > > > > > > > =================================================================================== > > > > = BAD TERMINATION OF ONE OF YOUR APPLICATION PROCESSES > > > > = PID 417219 RUNNING AT ssl_hn > > > > = EXIT CODE: 11 > > > > = CLEANING UP REMAINING PROCESSES > > > > = YOU CAN IGNORE THE BELOW CLEANUP MESSAGES > > > > > > > =================================================================================== > > > > Intel(R) MPI Library troubleshooting guide: > > > > https://software.intel.com/node/561764 > > > =================================================================================== > > > > > > > > For assimilation and dissemination of knowledge, visit cakes.cdac.in > > > > [image: 150th Anniversary Mahatma Gandhi] > > > > > ------------------------------------------------------------------------------------------------------------ > > > > [ C-DAC is on Social-Media too. Kindly follow us at: > > Facebook: https://www.facebook.com/CDACINDIA & Twitter: @cdacindia ] > > > > This e-mail is for the sole use of the intended recipient(s) and may > > contain confidential and privileged information. If you are not the > > intended recipient, please contact the sender by reply e-mail and destroy > > all copies and the original message. Any unauthorized review, use, > > disclosure, dissemination, forwarding, printing or copying of this email > > is strictly prohibited and appropriate legal action will be taken. > > > ------------------------------------------------------------------------------------------------------------ > > > > _______________________________________________ > > Quantum ESPRESSO is supported by MaX (www.max-centre.eu/quantum-espresso > ) > > users mailing list [email protected] > > https://lists.quantum-espresso.org/mailman/listinfo/users > > > > -- > Paolo Giannozzi, Dip. Scienze Matematiche Informatiche e Fisiche, > Univ. Udine, via delle Scienze 208, 33100 Udine, Italy > Phone +39-0432-558216, fax +39-0432-558222 > -------------- next part -------------- > An HTML attachment was scrubbed... > URL: < > http://lists.quantum-espresso.org/pipermail/users/attachments/20200121/55f06b9a/attachment-0001.html > > > -------------- next part -------------- > A non-text attachment was scrubbed... > Name: signature.jpg > Type: image/jpeg > Size: 7789 bytes > Desc: not available > URL: < > http://lists.quantum-espresso.org/pipermail/users/attachments/20200121/55f06b9a/attachment-0001.jpg > > > > ------------------------------ > > Message: 2 > Date: Tue, 21 Jan 2020 17:19:32 -0300 > From: [email protected] > To: [email protected] > Subject: [QE-users] mod value in polarization calculation with Berry > phase > Message-ID: > <[email protected]> > Content-Type: text/plain; charset=utf-8; format=flowed; DelSp=Yes > > Dear users and developers, > > I calculated the electric polarization of two structures, Ca3Mn2O7 and > Ca3Ti2O7, and I need some help to make sense of the results, > particularly the polarization quantum. > > At the output file for Ca3Ti2O7, where QE summarizes the results, it > is printed a mod 2: > > TOTAL PHASE: -0.53697 (mod 2) > > In this case, the polarization quantum is twice what it should be > (using the analytical expression). For Ca3Mn2O7, it prints mod 1 and > the polarization is the same as the one calculated analytically. > > What is this mod and why this diference? What is the correct > polarization quantum of Ca3Ti2O7: the one printed by QE or this value > divided by 2? > > Thanks in advance for your help! > > Michel L. Marcondes > Post-doctoral Research Scientist > University of Sao Paulo > > > > ------------------------------ > > Message: 3 > Date: Wed, 22 Jan 2020 11:18:50 +0530 > From: Poonam Kaushik <[email protected]> > To: [email protected] > Subject: [QE-users] NiO band structure and density of states > Message-ID: > <CAESGd6m13h5H67B=MMGSC7jj= > [email protected]> > Content-Type: text/plain; charset="utf-8" > > I calculated the bandgap and density of states of NiO by using LDA+U but i > m not getting the same bandgap in the density of states and in > bandstructure also i m not sure this band structure is correct or not. I m > a new user in quantum espresso. I attached my input file and band structure > also. I m confused what can i do further? > > > > > > ------------------------------------------------------------------------------------------------- > Poonam Sharma > Research Scholar > Department of Physics > Indian Institute of Technology Bombay > Mumbai - 400076 > India. > -------------- next part -------------- > An HTML attachment was scrubbed... > URL: < > http://lists.quantum-espresso.org/pipermail/users/attachments/20200122/b6e792c2/attachment-0001.html > > > -------------- next part -------------- > A non-text attachment was scrubbed... > Name: NiO_dos_20k.png > Type: image/png > Size: 38200 bytes > Desc: not available > URL: < > http://lists.quantum-espresso.org/pipermail/users/attachments/20200122/b6e792c2/attachment-0002.png > > > -------------- next part -------------- > A non-text attachment was scrubbed... > Name: band structure.png > Type: image/png > Size: 75470 bytes > Desc: not available > URL: < > http://lists.quantum-espresso.org/pipermail/users/attachments/20200122/b6e792c2/attachment-0003.png > > > -------------- next part -------------- > A non-text attachment was scrubbed... > Name: nio.scf.in > Type: application/octet-stream > Size: 746 bytes > Desc: not available > URL: < > http://lists.quantum-espresso.org/pipermail/users/attachments/20200122/b6e792c2/attachment-0002.obj > > > -------------- next part -------------- > A non-text attachment was scrubbed... > Name: nio.nscf.in > Type: application/octet-stream > Size: 792 bytes > Desc: not available > URL: < > http://lists.quantum-espresso.org/pipermail/users/attachments/20200122/b6e792c2/attachment-0003.obj > > > > ------------------------------ > > Message: 4 > Date: Wed, 22 Jan 2020 09:38:10 +0000 > From: Timrov Iurii <[email protected]> > To: "[email protected]" > <[email protected]> > Subject: Re: [QE-users] NiO band structure and density of states > Message-ID: <[email protected]> > Content-Type: text/plain; charset="us-ascii" > > Dear Poonam Sharma, > > > > ...i m not sure this band structure is correct or not. > > > Check the literature, there are many studies of NiO using DFT+U. For > example, see Fig. 2(b) in Campo and Cococcioni, J. Phys.: Condens. Matter > 22, 055602 (2010). You can try to use exactly the same parameters of > calculations as in that paper and try to reproduce Fig. 2(b). > > Note also that in the latest versions of Quantum ESPRESSO there is a code > (the HP code, hp.x) which can be used to compute Hubbard U from first > principles. > > The broadening parameter 0.05 Ry is large, I think you should reduce it. > For magnetic insulators I suggest to use the procedure described in > q-e/HP/examples/example02/README. > > > > I m a new user in quantum espresso > > > There are many tutorials based on Quantum ESPRESSO, it would be very > useful to check them: > https://www.quantum-espresso.org/news-events/complete-qe-schools-workshops-and-tutorials > > > HTH > > > Cheers, > > Iurii > > > -- > Dr. Iurii Timrov > Postdoctoral Researcher > STI - IMX - THEOS and NCCR - MARVEL > Swiss Federal Institute of Technology Lausanne (EPFL) > CH-1015 Lausanne, Switzerland > +41 21 69 34 881 > http://people.epfl.ch/265334 > ________________________________ > From: users <[email protected]> on behalf of > Poonam Kaushik <[email protected]> > Sent: Wednesday, January 22, 2020 6:48:50 AM > To: [email protected] > Subject: [QE-users] NiO band structure and density of states > > I calculated the bandgap and density of states of NiO by using LDA+U but > i m not getting the same bandgap in the density of states and in > bandstructure also i m not sure this band structure is correct or not. I m > a new user in quantum espresso. I attached my input file and band structure > also. I m confused what can i do further? > > > > > > ------------------------------------------------------------------------------------------------- > Poonam Sharma > Research Scholar > Department of Physics > Indian Institute of Technology Bombay > Mumbai - 400076 > India. > > -------------- next part -------------- > An HTML attachment was scrubbed... > URL: < > http://lists.quantum-espresso.org/pipermail/users/attachments/20200122/c9540163/attachment-0001.html > > > > ------------------------------ > > Subject: Digest Footer > > _______________________________________________ > users mailing list > [email protected] > https://lists.quantum-espresso.org/mailman/listinfo/users > > ------------------------------ > > End of users Digest, Vol 150, Issue 21 > ************************************** >
{kind=link}
{kind=link}
{kind=link}
_______________________________________________ Quantum ESPRESSO is supported by MaX (www.max-centre.eu/quantum-espresso) users mailing list [email protected] https://lists.quantum-espresso.org/mailman/listinfo/users