Hi,
The automatic procedure of relativistic splittings is implemented
properly only in very simple cases.
In particular, when the magnetization direction is changing, one would
need different linear combinations of l+m+s.
In principle you can use option 6 and provide a case.cf1 file, where the
proper linear combination is given manually, but this might need an
expert in relativity. (see the example case.cf* files in SRC_template)
At the moment, the only tip besides what I wrote above is that you try
to setup your system by interchanging the a and c axis for the (100)
case, such that M points again in (001) direction. But even then, I'm
not sure if the outcome will be correct.
Regards
On 06/23/2017 11:12 AM, Jindrich Kolorenc wrote:
Hello,
Thanks a lot for the updated files. Unfortunately, the fix appears to
work only partially:
1) total f DOS from "qtl" and from "lapw2 -qtl" are now the same
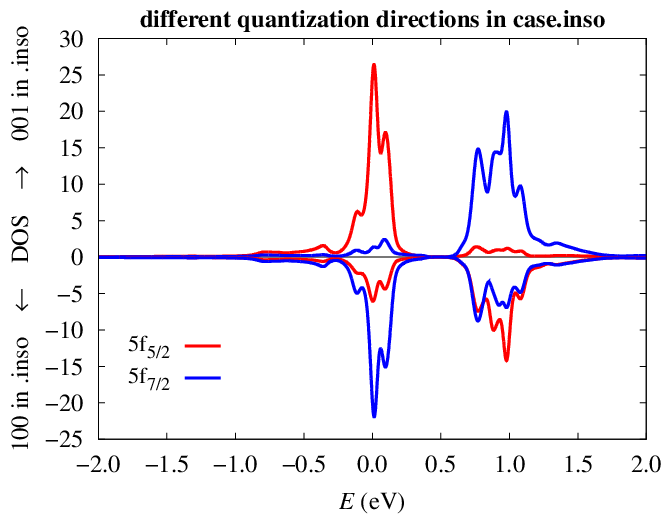
2) when the magnetization direction 001 is specified in case.inso, the
5/2 and 7/2 decomposition looks correct
The decomposition is still incorrect when the magnetization in case.inso
is set to 100, the lower-energy peak still comes out as dominantly 7/2:
http://www.fzu.cz/~kolorenc/wien2k_UGa2_bug/UGa2_f52_f72_001_100.png
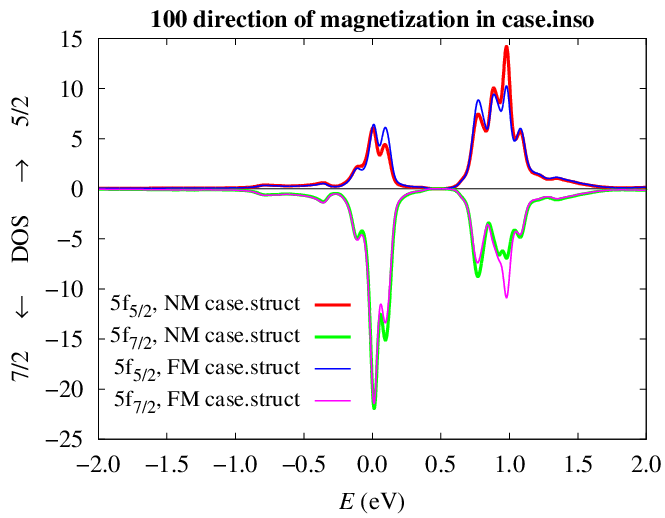
In the case of 100 direction, there are also small differences between
runs with the struct file I attached the last time (NM struct), and with
the struct file that is attached now (FM struct with a reduced number of
symmetry operations)
http://www.fzu.cz/~kolorenc/wien2k_UGa2_bug/UGa2_f52_f72_100.png
These differences show up only in the decomposed f DOS. The total f
DOSes are the same and so is the total energy (the difference is smaller
than 10e-6 Ry).
I have also tried calculations with U as atom number 1 in the struct
file, but it gives the same incorrect result for 5/2 and 7/2
decomposition in the 100 case.
Best regards,
Jindrich
PS: The reason why I look at the 100 direction is that my final goal is
the analysis of 100 ferromagnet.
On 06/22/2017 10:04 AM, Peter Blaha wrote:
Hi,
Thank you very much for this bug report. I can confirm the problem.
It was a nasty bug, occurring only if you have
. non-spinpolarized spin-orbit calculations AND
. more than one atom/cell (the first atom is always ok).
The calculated partial charges (case.qtl) are all wrong except for the
first atom (not only the relativistic decomposition into 5/2 and 7/2,
..., but also the regular partial charges using "x qtl".
In addition there was a factor of two in the normalization, so that the
total DOS could be less than the partial one.
I include a few subroutines for SRC_qtl which should fix the problem.
Please verify.
Regards
Peter Blaha
_______________________________________________
Wien mailing list
[email protected]
http://zeus.theochem.tuwien.ac.at/mailman/listinfo/wien
SEARCH the MAILING-LIST at:
http://www.mail-archive.com/[email protected]/index.html
--
P.Blaha
--------------------------------------------------------------------------
Peter BLAHA, Inst.f. Materials Chemistry, TU Vienna, A-1060 Vienna
Phone: +43-1-58801-165300 FAX: +43-1-58801-165982
Email: [email protected] WIEN2k: http://www.wien2k.at
WWW: http://www.imc.tuwien.ac.at/TC_Blaha
--------------------------------------------------------------------------
_______________________________________________
Wien mailing list
[email protected]
http://zeus.theochem.tuwien.ac.at/mailman/listinfo/wien
SEARCH the MAILING-LIST at:
http://www.mail-archive.com/[email protected]/index.html
{kind=link}
{kind=link}