V. Nagarajan wrote:
Is there some sort of consensus on what properties make a crystal diffract
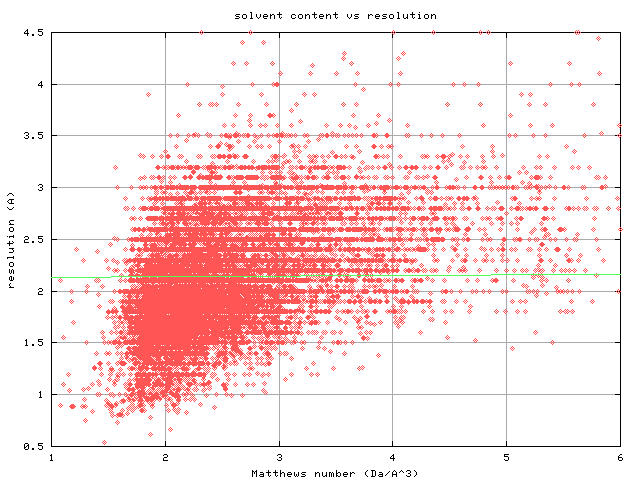
well or poorly? For example, solvent content is assumed to be critical.
Assumed? Yes. Critical? No. Bernhard Rupp will probably jump on me for
this, but there are plenty of high solvent content crystals that
diffract very well, and also plenty of low solvent content crystals that
don't diffract at all. There is a SLIGHT correlation between solvent
content and resolution in the PDB:
http://bl831.als.lbl.gov/~jamesh/Vm_vs_resolution.jpg
but the relationship is not what I would call "predictive". Solvent
content, it seems, is just one of the many straws people grasp in an
effort to explain why their crystals suck.
What
else?
Diffraction is much easier to understand when you realize that a
"crystal lattice" is just something we humans made up to make the math
easier. The molecules don't care about it. So, for ANY arrangement of
atoms, you can still declare an average lattice spacing simply by
dividing the size of the first Fresnel zone (sometimes called a
"coherence length") by the number of molecules that lie across it. For
example, with 1 A radiation and a detector at 100 mm from the sample the
first Fresnel zone is about 1 micron wide. Remember, you can make up any
lattice you like, even if the substance is completely amorphous.
The next step is to (somehow) take the electron density in each of
the unit cells that you have arbitrarily declared and then average them
all. The Fourier transform of this average electron density gives you
"F"s, and the squares of these are then proportional to spot intensities
(after correcting for Lorentz, polarization, and a few other effects).
It really is that simple. However, if your unit cell has nothing to do
with any real repeats in the substance, then averaging a million or so
out-of-register molecules will give you a flat average electron density,
and then all the Fourier terms (except F000) will be zero. This means
no spots.
Now, no spots does not mean no scattering. The number of photons
scattered by a given number of atoms is fixed, but photons that don't
contribute to spots contribute to the "background" (such as SAXS).
Can crystals even grow if there is short-range (less than beam radius)
disorder?
I'm not sure what you mean by "beam radius", but the sad truth is that
"crystals" can be effectively amorphous. It can be as blatant as
costume jewelry (where the stones have facets, but are made of glass or
plastic) or as subtle as a bend in an otherwise perfect lattice.
The key point is that unit cells need not be adjacent to one another for
their scattering to interact. Indeed, they can be up to a micron or
more apart! It is not hard to imagine how something as soft as a
protein crystal could deflect by ~4 A over this distance (1 micron is
10,000 A). In fact, it is somewhat astonishing how so many protein
crystal lattices are "straight" to within a few A over a micron,
especially after being pried off a piece of glass and beaten with a
nylon loop before being squeezed into the surface tension of a droplet
of liquid and then dunked into liquid nitrogen.
Nevertheless, the unfortunate truth about scattering physics is that
our imaginary "crystal lattice" which we must use to compute the average
electron density in a unit cell is PERFECTLY straight. You can "fit"
the imaginary lattice to the true repeating structure in your sample
(this is done by programs like DENZO and MOSFLM when they refine the
unit cell and crystal orientation), but any deviation of the "real"
lattice from this perfection over the entire Fresnel zone will lead to a
smearing of the average density. Blurry average density has weak
high-resolution Fourier terms, and it is also very difficult to fit a
single-conformer molecular model into it, even if you do apply a custom
Gaussian blurring filter (B factor) to each atom.
In this light, it is perhaps apparent how silly it is to be using a
model Debye, Waller and Ott derived to account for small thermal
vibrations to explain the kind of disorder we see in a soft, pliable
lattice at 100 Kelvin. Instead of a Gaussian on each atom, perhaps some
other function would be more appropriate? Something that reflects
correlated motions? Small wonder perhaps that TLS has been so successful?
Something I rediscovered recently is that the way an otherwise perfect
crystal lattice bends and stretches in response to a defect was worked
out over 50 years ago by H. Kanzaki (1957) J. Phys. Chem. Solids. vol 2.
pp. 24-36, who spawned what is still an active field of research in how
dopants and other defects change the crystal lattice of silicon and
other commercially important crystals. I think the only reason this
"Kanzaki force" formalism is not used in protein crystallography is
because the equations are too complicated for simple-minded biologists
(such as myself) to comprehend.
-James Holton
MAD Scientist
Thanks,
V. Nagarajan
JAN Scientific, Inc.
http://janscientific.com
-----Original Message-----
From: CCP4 bulletin board [mailto:[email protected]] On Behalf Of James
Holton
Sent: Thursday, June 18, 2009 11:04 AM
To: [email protected]
Subject: Re: [ccp4bb] Phantom Crystals
[deleted]
Nevertheless, I think it is still up in the air how much diffraction
tends to be degraded by crystal handling vs crystals just being "born
ugly", as the proper control (shooting crystals without handling them)
has not been done on anything but a few test cases. In fact, I have
heard enough stories about ugly crystals diffracting very well and
beautiful crystals diffracting poorly to wonder if these two qualities
really are anticorellated. That is, beauty really is just "skin deep"
(and ugly goes to the core). I think it will be telling to see what sort
of results we get from the now several available "in-situ" diffraction
systems <shameless plug>one of which myself and others developed with
Fluidigm, who are now selling them</shameless plug>.
-James Holton
MAD Scientist
{kind=link}