There are 13 messages totalling 1026 lines in this issue.
Topics of the day:
1. refmac failed message (2)
2. question of extra high B factor (6)
3. Coot:findwaters in REFMAC (2)
4. Postdoctoral position in protein crystallography at Karolinska
Institutet
5. OpenGL Stereo 3D on 120 Hz LCDs, at last!
6. Foils for energy calibration
----------------------------------------------------------------------
Date: Thu, 30 Jul 2009 10:50:13 GMT
From: Elad Binshtien <[email protected]>
Subject: refmac failed message
This is a multi-part message in MIME format.
----8d5c55134e491a1d1bdd
Content-Type: text/plain; charset=utf-8
Content-Disposition: inline
Content-Transfer-Encoding: quoted-printable
Dear all=2C
I am refining a structure in Refmac at 2=2E2 A in win OS=2E=C2=A0
Howeve=
r=2C
Refmac failed and send this message=3A =C2=A0 =C2=A0 =C2=A0=C2=A0
forrtl=
=3A error (72)=3A floating overflow=C2=A0 =
Thank you in advance for your any helpful suggestions=2E=C2=A0 =
Best=2C
Elad
Elad Binshtein
Ph=2ED student =
Department of Life Science =
Ben Gurion University of the Negev
Ph=3A 972-8-6461325=E2=80=8E
----8d5c55134e491a1d1bdd
Content-Type: text/html; charset=utf-8
Content-Disposition: inline
Content-Transfer-Encoding: quoted-printable
Dear all=2C=3Cbr=3E=3Cbr=3E=3Cbr=3EI am refining a structure in
Refmac a=
t 2=2E2 A in win OS=2E=26nbsp=3B However=2C
Refmac failed and send this message=3A =26nbsp=3B =26nbsp=3B
=26nbsp=3B=26=
nbsp=3B forrtl=3A error (72)=3A floating overflow=26nbsp=3B
=3Cbr=3E=3Cb=
r=3E Thank you in advance for your any helpful
suggestions=2E=26nbsp=3B =
=
3Cbr
=3E=3Cbr=3E=3Cbr=3EBest=2C=3Cbr=3EElad=3Cbr=3E=3CBR=3E=3CBR=3EElad =
Binshtein=3Cbr=3EPh=2ED student =3Cbr=3EDepartment of Life Science
=3Cbr=
=3EBen Gurion University of the Negev=3Cbr=3EPh=3A 972-8-6461325=3C/
BR=3E=
=3C/BR=3E=E2=80=8E
----8d5c55134e491a1d1bdd--
------------------------------
Date: Thu, 30 Jul 2009 13:25:43 +0100
From: "Stein, ND (Norman)" <[email protected]>
Subject: Re: refmac failed message
This is a multi-part message in MIME format.
------_=_NextPart_001_01CA1110.DB7CC0CB
Content-Type: text/plain;
charset="windows-1255"
Content-Transfer-Encoding: quoted-printable
Hi Elad
=20
You don't say which version of Refmac you are using but I think the =
first thing to do would be to try the latest version (5.5.0102). You
can =
pick this up from
=20
ftp://ftp.ccp4.ac.uk/nds/windows/refmac_5.5.0102/refmac5.exe
=20
Copy this exe file to the bin subdirectory of your CCP4
installation. =
(Probably wise to make a backup copy of the existing refmac5.exe
first).
=20
Best wishes
=20
Norman Stein
CCP4
________________________________
From: CCP4 bulletin board [mailto:[email protected]] On Behalf
Of =
Elad Binshtien
Sent: 30 July 2009 11:50
To: [email protected]
Subject: [ccp4bb] refmac failed message
Dear all,
I am refining a structure in Refmac at 2.2 A in win OS. However,
Refmac =
failed and send this message: forrtl: error (72): floating =
overflow =20
Thank you in advance for your any helpful suggestions. =20
Best,
Elad
Elad Binshtein
Ph.D student=20
Department of Life Science=20
Ben Gurion University of the Negev
Ph: 972-8-6461325
=FD=20
-- =0AScanned by iCritical.=0A
------_=_NextPart_001_01CA1110.DB7CC0CB
Content-Type: text/html;
charset="windows-1255"
Content-Transfer-Encoding: quoted-printable
<!DOCTYPE HTML PUBLIC "-//W3C//DTD HTML 4.0 Transitional//EN">
<HTML><HEAD>
<META http-equiv=3DContent-Type content=3D"text/html; =
charset=3Dwindows-1255">
<META content=3D"MSHTML 6.00.2900.3562" name=3DGENERATOR></HEAD>
<BODY>
<DIV dir=3Dltr align=3Dleft><SPAN class=3D419481712-30072009><FONT =
face=3DArial=20
color=3D#0000ff size=3D2>Hi Elad</FONT></SPAN></DIV>
<DIV dir=3Dltr align=3Dleft><SPAN class=3D419481712-30072009><FONT =
face=3DArial=20
color=3D#0000ff size=3D2></FONT></SPAN> </DIV>
<DIV dir=3Dltr align=3Dleft><SPAN class=3D419481712-30072009><FONT =
face=3DArial=20
color=3D#0000ff size=3D2>You don't say which version of Refmac you
are =
using but I=20
think the first thing to do would be to try the latest =
version (5.5.0102).=20
You can pick this up from</FONT></SPAN></DIV>
<DIV dir=3Dltr align=3Dleft><SPAN class=3D419481712-30072009><FONT =
face=3DArial=20
color=3D#0000ff size=3D2></FONT></SPAN> </DIV>
<DIV dir=3Dltr align=3Dleft><SPAN class=3D419481712-30072009><FONT =
face=3DArial=20
color=3D#0000ff size=3D2><A=20
href=3D"ftp://ftp.ccp4.ac.uk/nds/windows/refmac_5.5.0102/
refmac5.exe">ftp=
://ftp.ccp4.ac.uk/nds/windows/refmac_5.5.0102/refmac5.exe</A></
FONT></SPA=
N></DIV>
<DIV dir=3Dltr align=3Dleft><SPAN class=3D419481712-30072009><FONT =
face=3DArial=20
color=3D#0000ff size=3D2></FONT></SPAN> </DIV>
<DIV dir=3Dltr align=3Dleft><SPAN class=3D419481712-30072009><FONT =
face=3DArial=20
color=3D#0000ff size=3D2>Copy this exe file to the bin subdirectory
of=20
your CCP4 installation. (Probably wise to make a backup
copy =
of the=20
existing refmac5.exe first).</FONT></SPAN></DIV>
<DIV dir=3Dltr align=3Dleft><SPAN class=3D419481712-30072009><FONT =
face=3DArial=20
color=3D#0000ff size=3D2></FONT></SPAN> </DIV>
<DIV dir=3Dltr align=3Dleft><SPAN class=3D419481712-30072009><FONT =
face=3DArial=20
color=3D#0000ff size=3D2>Best wishes</FONT></SPAN></DIV>
<DIV dir=3Dltr align=3Dleft><SPAN class=3D419481712-30072009><FONT =
face=3DArial=20
color=3D#0000ff size=3D2></FONT></SPAN> </DIV>
<DIV dir=3Dltr align=3Dleft><SPAN class=3D419481712-30072009><FONT =
face=3DArial=20
color=3D#0000ff size=3D2>Norman Stein</FONT></SPAN></DIV>
<DIV dir=3Dltr align=3Dleft><SPAN class=3D419481712-30072009><FONT =
face=3DArial=20
color=3D#0000ff size=3D2>CCP4</FONT></SPAN></DIV><BR>
<DIV class=3DOutlookMessageHeader lang=3Den-us dir=3Dltr align=3Dleft>
<HR tabIndex=3D-1>
<FONT face=3DTahoma size=3D2><B>From:</B> CCP4 bulletin board=20
[mailto:[email protected]] <B>On Behalf Of </B>Elad=20
Binshtien<BR><B>Sent:</B> 30 July 2009 11:50<BR><B>To:</B>=20
[email protected]<BR><B>Subject:</B> [ccp4bb] refmac failed=20
message<BR></FONT><BR></DIV>
<DIV></DIV>Dear all,<BR><BR><BR>I am refining a structure in Refmac
at =
2.2 A in=20
win OS. However, Refmac failed and send this message: =
=20
forrtl: error (72): floating overflow
<BR><BR>Thank =
you in=20
advance for your any helpful suggestions. =20
<BR><BR><BR>Best,<BR>Elad<BR><BR><BR>Elad Binshtein<BR>Ph.D student=20
<BR>Department of Life Science <BR>Ben Gurion University of the =
Negev<BR>Ph:=20
972-8-6461325<BR><BR>=FD
<br>=
<p>-- =0A<BR>Scanned by iCritical.=0A</p>
<br>=
</BODY></HTML>
------_=_NextPart_001_01CA1110.DB7CC0CB--
------------------------------
Date: Fri, 31 Jul 2009 00:00:25 +0800
From: Jiamu Du <[email protected]>
Subject: question of extra high B factor
--00221504874fd6c69c046fee6655
Content-Type: text/plain; charset=ISO-8859-1
Content-Transfer-Encoding: 7bit
Dear All,
I am refining a structure of a complex between of 50kD protein and a
20kD
glycosylated protien. The data is of 2.9 A resolution. The wilson B
factor
is as high as 86.3 A^2.
The refinement seems well with R/Rf of 0.21/0.25. But the B-factor
is extra
high. For the 50kD part, the average B factor is 76.5 A^2. But the B
factor
of the 20 kD glycosylated protein is as high as 133.3 A^2. Although
the
electron density looks fine, even the sugar chain is seen clearly.
My question is:
1. How to reduce the B factor to a reasonable level?
2. If it can not be redueced, when I published it, is this value
acceptable?
3. In the same of similar resolutionIs, is there some other
structures like
this situation? A component or a subunit of the protein has a extra
high B
factor as high as 130.
Thanks and best wishes.
--
Jiamu Du, Ph.D.
State Key Laboratory of Molecular Biology
Institute of Biochemistry and Cell Biology
Shanghai Institutes for Biological Sciences
Chinese Academy of Sciences
320 Yue-Yang Road
Shanghai 200031
P. R. China
Tel: +86-21-5492-1117
E-mail: [email protected]
--00221504874fd6c69c046fee6655
Content-Type: text/html; charset=ISO-8859-1
Content-Transfer-Encoding: quoted-printable
Dear All,<br>I am refining a structure of a complex between of 50kD
protein=
and a 20kD glycosylated protien. The data is of 2.9 A resolution.
The wils=
on B factor is as high as 86.3 A^2.<br>The refinement seems well
with R/Rf =
of 0.21/0.25. But the B-factor is extra high. For the 50kD part, the
averag=
e B factor is 76.5 A^2. But the B factor of the 20 kD glycosylated
protein =
is as high as 133.3 A^2. Although the electron density looks fine,
even the=
sugar chain is seen clearly.<br>
My question is:<br>1. How to reduce the B factor to a reasonable
level?<br>=
2. If it can not be redueced, when I published it, is this value
acceptable=
?<br>3. In the same of similar resolutionIs, is there some other
structures=
like this situation?=A0 A component or a subunit of the protein has
a extr=
a high B factor as high as 130.<br>
<br>Thanks and best wishes.<br><br clear=3D"all"><br>-- <br>Jiamu
Du, Ph.D.=
<br>State Key Laboratory of Molecular Biology<br>Institute of
Biochemistry =
and Cell Biology<br>Shanghai Institutes for Biological
Sciences<br>Chinese =
Academy of Sciences<br>
320 Yue-Yang Road<br>Shanghai 200031<br>P. R. China<br>Tel:
+86-21-5492-111=
7<br>E-mail: <a href=3D"mailto:[email protected]";>[email protected]</
a><br>
--00221504874fd6c69c046fee6655--
------------------------------
Date: Thu, 30 Jul 2009 09:32:26 -0700
From: Pavel Afonine <[email protected]>
Subject: Re: question of extra high B factor
Hi Jiamu,
My question is:
1. How to reduce the B factor to a reasonable level?
3. In the same of similar resolutionIs, is there some other
structures
like this situation?
POLYGON tool is (one of) your friend(s) to answer this question (apart
from debatable one about "a reasonable level"):
Acta Cryst. D65, 297-300 (2009).
Now it is available in PHENIX (from the latest nightly builds):
http://www.phenix-online.org/
Please let me know if you have any questions about it.
Pavel.
------------------------------
Date: Thu, 30 Jul 2009 17:47:55 +0100
From: Clemens Vonrhein <[email protected]>
Subject: Re: question of extra high B factor
Dear Jiamu,
On Fri, Jul 31, 2009 at 12:00:25AM +0800, Jiamu Du wrote:
I am refining a structure of a complex between of 50kD protein and
a 20kD
glycosylated protien. The data is of 2.9 A resolution. The wilson B
factor
is as high as 86.3 A^2.
The refinement seems well with R/Rf of 0.21/0.25. But the B-factor
is extra
high. For the 50kD part, the average B factor is 76.5 A^2. But the
B factor
of the 20 kD glycosylated protein is as high as 133.3 A^2. Although
the
electron density looks fine, even the sugar chain is seen clearly.
My question is:
1. How to reduce the B factor to a reasonable level?
How do you define 'reasonable'? And why would you want to reduce this
anyway?
( 50*76.5 + 20*133.3 ) / 70 = 92.7
which seems fairly close to the Wilson B of 86.3, right?
2. If it can not be redueced, when I published it, is this value
acceptable?
Better question: is it the right value? Remember it is
results -> publish
and not
publish <- results
;-)
If they're correct than they are acceptable (I would accept those
values).
3. In the same of similar resolutionIs, is there some other
structures like
this situation? A component or a subunit of the protein has a
extra high B
factor as high as 130.
I'm sure hundreds of them ... but I'm sure you want your structure to
stand on its own, so don't look too close at other structures and
repeat the various mistakes we've all made and that are now set in
stone in some old PDB file.
Cheers
Clemens
--
***************************************************************
* Clemens Vonrhein, Ph.D. vonrhein AT GlobalPhasing DOT com
*
* Global Phasing Ltd.
* Sheraton House, Castle Park
* Cambridge CB3 0AX, UK
*--------------------------------------------------------------
* BUSTER Development Group (http://www.globalphasing.com)
***************************************************************
------------------------------
Date: Fri, 31 Jul 2009 00:02:42 +0800
From: Jiamu Du <[email protected]>
Subject: Re: question of extra high B factor
--0022152d604d09ee86046fee6f07
Content-Type: text/plain; charset=ISO-8859-1
Content-Transfer-Encoding: 7bit
I am using TLS refinement with Phenix for the structure refinement.
On Fri, Jul 31, 2009 at 12:00 AM, Jiamu Du <[email protected]> wrote:
Dear All,
I am refining a structure of a complex between of 50kD protein and
a 20kD
glycosylated protien. The data is of 2.9 A resolution. The wilson B
factor
is as high as 86.3 A^2.
The refinement seems well with R/Rf of 0.21/0.25. But the B-factor
is extra
high. For the 50kD part, the average B factor is 76.5 A^2. But the
B factor
of the 20 kD glycosylated protein is as high as 133.3 A^2. Although
the
electron density looks fine, even the sugar chain is seen clearly.
My question is:
1. How to reduce the B factor to a reasonable level?
2. If it can not be redueced, when I published it, is this value
acceptable?
3. In the same of similar resolutionIs, is there some other
structures like
this situation? A component or a subunit of the protein has a
extra high B
factor as high as 130.
Thanks and best wishes.
--
Jiamu Du, Ph.D.
State Key Laboratory of Molecular Biology
Institute of Biochemistry and Cell Biology
Shanghai Institutes for Biological Sciences
Chinese Academy of Sciences
320 Yue-Yang Road
Shanghai 200031
P. R. China
Tel: +86-21-5492-1117
E-mail: [email protected]
--
Jiamu Du, Ph.D.
State Key Laboratory of Molecular Biology
Institute of Biochemistry and Cell Biology
Shanghai Institutes for Biological Sciences
Chinese Academy of Sciences
320 Yue-Yang Road
Shanghai 200031
P. R. China
Tel: +86-21-5492-1117
E-mail: [email protected]
--0022152d604d09ee86046fee6f07
Content-Type: text/html; charset=ISO-8859-1
Content-Transfer-Encoding: quoted-printable
I am using TLS refinement with Phenix for the structure
refinement.<br><br>=
<br><div class=3D"gmail_quote">On Fri, Jul 31, 2009 at 12:00 AM,
Jiamu Du <=
span dir=3D"ltr"><<a href=3D"mailto:[email protected]";>[email protected]
=
</a>></span> wrote:<br>
<blockquote class=3D"gmail_quote" style=3D"border-left: 1px solid
rgb(204, =
204, 204); margin: 0pt 0pt 0pt 0.8ex; padding-left: 1ex;">Dear
All,<br>I am=
refining a structure of a complex between of 50kD protein and a 20kD
glyco=
sylated protien. The data is of 2.9 A resolution. The wilson B
factor is as=
high as 86.3 A^2.<br>
The refinement seems well with R/Rf of 0.21/0.25. But the B-factor
is extra=
high. For the 50kD part, the average B factor is 76.5 A^2. But the B
facto=
r of the 20 kD glycosylated protein is as high as 133.3 A^2.
Although the e=
lectron density looks fine, even the sugar chain is seen clearly.<br>
My question is:<br>1. How to reduce the B factor to a reasonable
level?<br>=
2. If it can not be redueced, when I published it, is this value
acceptable=
?<br>3. In the same of similar resolutionIs, is there some other
structures=
like this situation?=A0 A component or a subunit of the protein has
a extr=
a high B factor as high as 130.<br>
<br>Thanks and best wishes.<br><br clear=3D"all"><br>-- <br>Jiamu
Du, Ph.D.=
<br>State Key Laboratory of Molecular Biology<br>Institute of
Biochemistry =
and Cell Biology<br>Shanghai Institutes for Biological
Sciences<br>Chinese =
Academy of Sciences<br>
320 Yue-Yang Road<br>Shanghai 200031<br>P. R. China<br>Tel:
+86-21-5492-111=
7<br>E-mail: <a href=3D"mailto:[email protected]";
target=3D"_blank">jiamudu=
@gmail.com</a><br>
</blockquote></div><br><br clear=3D"all"><br>-- <br>Jiamu Du,
Ph.D.<br>Stat=
e Key Laboratory of Molecular Biology<br>Institute of Biochemistry
and Cell=
Biology<br>Shanghai Institutes for Biological Sciences<br>Chinese
Academy =
of Sciences<br>
320 Yue-Yang Road<br>Shanghai 200031<br>P. R. China<br>Tel:
+86-21-5492-111=
7<br>E-mail: <a href=3D"mailto:[email protected]";>[email protected]</
a><br>
--0022152d604d09ee86046fee6f07--
------------------------------
Date: Thu, 30 Jul 2009 10:24:25 -0700
From: Huiying Li <[email protected]>
Subject: Coot:findwaters in REFMAC
I used Coot:findwaters facility in REFMAC (CCP4 6.1.1) to add waters
to
the refined structure. Often for the well ordered water sites the
routine added two water molecules in single water site, with their
distance (~1 Angs) way shorter than allowed hydrogen bonding
distance. I
have to remove the extra water molecules manually.
Is this a intended feature? It seems to only create extra work to the
user. What is the purpose of having different chain IDs for waters
output
from this routine?
Huiying Li, Ph. D
Department of Molecular Biology and Biochemistry
Natural Sciences I, Rm 2443
University of California at Irvine
Irvine, CA 92697, USA
Tel: 949-824-4322(or -1953); Fax: 949-824-3280
email: [email protected]
------------------------------
Date: Thu, 30 Jul 2009 18:33:01 +0100
From: Paul Emsley <[email protected]>
Subject: Re: Coot:findwaters in REFMAC
Huiying Li wrote:
I used Coot:findwaters facility in REFMAC (CCP4 6.1.1) to add waters
to the refined structure. Often for the well ordered water sites the
routine added two water molecules in single water site, with their
distance (~1 Angs) way shorter than allowed hydrogen bonding
distance.
I have to remove the extra water molecules manually.
Sounds like an old version. Things have improved.
Is this a intended feature? It seems to only create extra work to the
user.
:-(
What is the purpose of having different chain IDs for waters output
from this routine?
I thought (at the time) that it was sensible to add waters to a
different chain ID to the protein atoms (I still do, in fact). There
are others (somewhat less involved in model-building I suspect) that
think otherwise.
Paul.
------------------------------
Date: Thu, 30 Jul 2009 20:56:14 +0200
From: Kay Diederichs <[email protected]>
Subject: Re: question of extra high B factor
This is a cryptographically signed message in MIME format.
--------------ms000208020902090005010407
Content-Type: text/plain; charset=ISO-8859-1; format=flowed
Content-Transfer-Encoding: 7bit
Jiamu Du schrieb:
Dear All,
I am refining a structure of a complex between of 50kD protein and a
20kD glycosylated protien. The data is of 2.9 A resolution. The
wilson B
factor is as high as 86.3 A^2.
The refinement seems well with R/Rf of 0.21/0.25. But the B-factor is
extra high. For the 50kD part, the average B factor is 76.5 A^2.
But the
B factor of the 20 kD glycosylated protein is as high as 133.3 A^2.
Although the electron density looks fine, even the sugar chain is
seen
clearly.
My question is:
1. How to reduce the B factor to a reasonable level?
Please check out
<http://strucbio.biologie.uni-konstanz.de/ccp4wiki/index.php/Refinement#help.2C_my_protein_has_high_B-factors.21
>
2. If it can not be redueced, when I published it, is this value
acceptable?
As there's nothing wrong with it, it can be published.
3. In the same of similar resolutionIs, is there some other
structures
like this situation? A component or a subunit of the protein has a
extra high B factor as high as 130.
Just look at the B-factors of new PDB entries which have a a
resolution
worse than 3 A (e.g., membrane proteins) - there are quite a few of
those.
HTH,
Kay
--
Kay Diederichs http://strucbio.biologie.uni-
konstanz.de
email: [email protected] Tel +49 7531 88 4049 Fax
3183
Fachbereich Biologie, Universitaet Konstanz, Box M647, D-78457
Konstanz
--------------ms000208020902090005010407
Content-Type: application/x-pkcs7-signature; name="smime.p7s"
Content-Transfer-Encoding: base64
Content-Disposition: attachment; filename="smime.p7s"
Content-Description: S/MIME Cryptographic Signature
MIAGCSqGSIb3DQEHAqCAMIACAQExCzAJBgUrDgMCGgUAMIAGCSqGSIb3DQEHAQAAoIIOlTCC
BM8wggQ4oAMCAQICDwCUsAABAAJijbUfbuxyZDANBgkqhkiG9w0BAQUFADCBvDELMAkGA1UE
BhMCREUxEDAOBgNVBAgTB0hhbWJ1cmcxEDAOBgNVBAcTB0hhbWJ1cmcxOjA4BgNVBAoTMVRD
IFRydXN0Q2VudGVyIGZvciBTZWN1cml0eSBpbiBEYXRhIE5ldHdvcmtzIEdtYkgxIjAgBgNV
BAsTGVRDIFRydXN0Q2VudGVyIENsYXNzIDEgQ0ExKTAnBgkqhkiG9w0BCQEWGmNlcnRpZmlj
YXRlQHRydXN0Y2VudGVyLmRlMB4XDTA4MDcxODExMzg1NFoXDTEwMTIzMTIyNTk1OVowfDEL
MAkGA1UEBhMCREUxHDAaBgNVBAoTE1RDIFRydXN0Q2VudGVyIEdtYkgxJTAjBgNVBAsTHFRD
IFRydXN0Q2VudGVyIENsYXNzIDEgTDEgQ0ExKDAmBgNVBAMTH1RDIFRydXN0Q2VudGVyIENs
YXNzIDEgTDEgQ0EgVkkwgZ8wDQYJKoZIhvcNAQEBBQADgY0AMIGJAoGBAJQFP39OlsR
+FrO6
sIC5tx+k3aTjsL4oCLrsrd2Z9nVMzrHxnvShlmxLhgElFThIoRzo7gr1mZbx2/
Gu3X51SEBN
TkP3bV2U0jn9yxGo97oYWA/+cRUKEEsxfvbPX5DL992EupcEptQAVutm32so3dGi
+nqe2mFX
wx8wpDAnCkDRAgMBAAGjggIQMIICDDCBjgYIKwYBBQUHAQEEgYEwfzBMBggrBgEFBQcwAoZA
aHR0cDovL3d3dy50cnVzdGNlbnRlci5kZS9jZXJ0c2VydmljZXMvY2FjZXJ0cy90Y19jbGFz
c18xX2NhLmNydDAvBggrBgEFBQcwAYYjaHR0cDovL29jc3AudGNjbGFzczEudHJ1c3RjZW50
ZXIuZGUwDwYDVR0TAQH/BAUwAwEB/
zBKBgNVHSAEQzBBMD8GCSqCFAAsAQEBATAyMDAGCCsG
AQUFBwIBFiRodHRwOi8vd3d3LnRydXN0Y2VudGVyLmRlL2d1aWRlbGluZXMwDgYDVR0PAQH
/
BAQDAgGGMB0GA1UdDgQWBBROm/Wy+91qa9xKB6/Ju2T/
FksmnzCB7AYDVR0fBIHkMIHhMIHe
oIHboIHYhjtodHRwOi8vY3JsLnRjY2xhc3MxLnRydXN0Y2VudGVyLmRlL2NybC92Mi90Y19j
bGFzc18xX2NhLmNybIaBmGxkYXA6Ly93d3cudHJ1c3RjZW50ZXIuZGUvQ049VEMlMjBUcnVz
dENlbnRlciUyMENsYXNzJTIwMSUyMENBLE89VEMlMjBUcnVzdENlbnRlciUyMEFHLG91PXJv
b3RjZXJ0cyxkYz10cnVzdGNlbnRlcixkYz1kZT9jZXJ0aWZpY2F0ZVJldm9jYXRpb25MaXN0
P2Jhc2U/MA0GCSqGSIb3DQEBBQUAA4GBAG4ApLln3RLqDiymjdk8SFs8S6acQryrkZB/
DJ2Q
H6n+6LYqEjirh+/Zj8uweQL6p6d9g/dl4S1wpIGCVC33AdcM8qer/
Cn0qcQ6bdduesfTcdQv
5uZlSsyEdKNa44DVtOh3IkTLtFdcDiY4tQFriM7kVC5Xuhyfxo58V1vOM8r
+MIIE3TCCBEag
AwIBAgIPAMYpAAEAAi4JCW7o2XnMMA0GCSqGSIb3DQEBBQUAMHwxCzAJBgNVBAYTAkRFMRww
GgYDVQQKExNUQyBUcnVzdENlbnRlciBHbWJIMSUwIwYDVQQLExxUQyBUcnVzdENlbnRlciBD
bGFzcyAxIEwxIENBMSgwJgYDVQQDEx9UQyBUcnVzdENlbnRlciBDbGFzcyAxIEwxIENBIFZJ
MB4XDTA5MDYxMTA3MzM1NVoXDTEwMDYxMjA3MzM1NVowWTELMAkGA1UEBhMCREUxGzAZBgNV
BAMTEkRyLiBLYXkgRGllZGVyaWNoczEtMCsGCSqGSIb3DQEJARYea2F5LmRpZWRlcmljaHNA
dW5pLWtvbnN0YW56LmRlMIIBIjANBgkqhkiG9w0BAQEFAAOCAQ8AMIIBCgKCAQEAq8l
+Wnxi
cqMo9KicOd1kqdaq3pAymwWO/
J7ejbVjKChe10XnaITas8R1k9FrF54FsvssThDwmwsDWFjh
eE0Fyegt+fk2tpXX4YnjqUcrlHkO5YHGQ0ogcrhvaMEKYoKLriH/
Nip0o8ljtMesL33gSPeV
BOpyFwngqYUCt0rZPBytI+wxpN+SAEAO6erxvC4pASYpGTU5KBYPi5o5yf
+9damc3flRF6Jl
tbX+5aBFkra8CiWjH3q44DnnPn/DI+0HZs5goQWPmvzLA5GMy5T
+3oXCGOKNFYFp7Y2AFdDf
2NyqSAo5KSd1D5qhCGWVedTeot435M5lqYWPJDFJj8Ew5QIDAQABo4IB/
jCCAfowgZcGCCsG
AQUFBwEBBIGKMIGHMFEGCCsGAQUFBzAChkVodHRwOi8vd3d3LnRydXN0Y2VudGVyLmRlL2Nl
cnRzZXJ2aWNlcy9jYWNlcnRzL3RjX2NsYXNzMV9MMV9DQV9WSS5jcnQwMgYIKwYBBQUHMAGG
Jmh0dHA6Ly9vY3NwLlZJLnRjY2xhc3MxLnRydXN0Y2VudGVyLmRlMB8GA1UdIwQYMBaAFE6b
9bL73Wpr3EoHr8m7ZP8WSyafMAwGA1UdEwEB/wQCMAAwSgYDVR0gBEMwQTA/
BgkqghQALAEB
AQEwMjAwBggrBgEFBQcCARYkaHR0cDovL3d3dy50cnVzdGNlbnRlci5kZS9ndWlkZWxpbmVz
MA4GA1UdDwEB/wQEAwIE8DAdBgNVHQ4EFgQUac6i
+TKOEt89dgE1SZwkDGCIPEcwVAYDVR0f
BE0wSzBJoEegRYZDaHR0cDovL2NybC5WSS50Y2NsYXNzMS50cnVzdGNlbnRlci5kZS9jcmwv
djIvdGNfY2xhc3MxX0wxX0NBX1ZJLmNybDAzBgNVHSUELDAqBggrBgEFBQcDAgYIKwYBBQUH
AwQGCCsGAQUFBwMHBgorBgEEAYI3FAICMCkGA1UdEQQiMCCBHmtheS5kaWVkZXJpY2hzQHVu
aS1rb25zdGFuei5kZTANBgkqhkiG9w0BAQUFAAOBgQBxO9sd6s4Lh9a2S70xvqvPkMZE6im
+
kI61FLhAhJqOqY3okJuO6xIdMjgbwblAuoEkHRaDTSna/2clqwH1k/
PiHn0TLHwWRCLIW3iO
WjwkIewI1Jn3xsmxrvc3TLec69BcKHDwcsuE2rSv2GzM9qH0Vx7EuVehRrCa1oZNgQsEZzCC
BN0wggRGoAMCAQICDwDGKQABAAIuCQlu6Nl5zDANBgkqhkiG9w0BAQUFADB8MQswCQYDVQQG
EwJERTEcMBoGA1UEChMTVEMgVHJ1c3RDZW50ZXIgR21iSDElMCMGA1UECxMcVEMgVHJ1c3RD
ZW50ZXIgQ2xhc3MgMSBMMSBDQTEoMCYGA1UEAxMfVEMgVHJ1c3RDZW50ZXIgQ2xhc3MgMSBM
MSBDQSBWSTAeFw0wOTA2MTEwNzMzNTVaFw0xMDA2MTIwNzMzNTVaMFkxCzAJBgNVBAYTAkRF
MRswGQYDVQQDExJEci4gS2F5IERpZWRlcmljaHMxLTArBgkqhkiG9w0BCQEWHmtheS5kaWVk
ZXJpY2hzQHVuaS1rb25zdGFuei5kZTCCASIwDQYJKoZIhvcNAQEBBQADggEPADCCAQoCggEB
AKvJflp8YnKjKPSonDndZKnWqt6QMpsFjvye3o21YygoXtdF52iE2rPEdZPRaxeeBbL7LE4Q
8JsLA1hY4XhNBcnoLfn5NraV1+GJ46lHK5R5DuWBxkNKIHK4b2jBCmKCi64h/
zYqdKPJY7TH
rC994Ej3lQTqchcJ4KmFArdK2TwcrSPsMaTfkgBADunq8bwuKQEmKRk1OSgWD4uaOcn/
vXWp
nN35UReiZbW1/uWgRZK2vAolox96uOA55z5/wyPtB2bOYKEFj5r8ywORjMuU/
t6FwhjijRWB
ae2NgBXQ39jcqkgKOSkndQ+aoQhllXnU3qLeN+TOZamFjyQxSY/
BMOUCAwEAAaOCAf4wggH6
MIGXBggrBgEFBQcBAQSBijCBhzBRBggrBgEFBQcwAoZFaHR0cDovL3d3dy50cnVzdGNlbnRl
ci5kZS9jZXJ0c2VydmljZXMvY2FjZXJ0cy90Y19jbGFzczFfTDFfQ0FfVkkuY3J0MDIGCCsG
AQUFBzABhiZodHRwOi8vb2NzcC5WSS50Y2NsYXNzMS50cnVzdGNlbnRlci5kZTAfBgNVHSME
GDAWgBROm/Wy+91qa9xKB6/Ju2T/
FksmnzAMBgNVHRMBAf8EAjAAMEoGA1UdIARDMEEwPwYJ
KoIUACwBAQEBMDIwMAYIKwYBBQUHAgEWJGh0dHA6Ly93d3cudHJ1c3RjZW50ZXIuZGUvZ3Vp
ZGVsaW5lczAOBgNVHQ8BAf8EBAMCBPAwHQYDVR0OBBYEFGnOovkyjhLfPXYBNUmcJAxgiDxH
MFQGA1UdHwRNMEswSaBHoEWGQ2h0dHA6Ly9jcmwuVkkudGNjbGFzczEudHJ1c3RjZW50ZXIu
ZGUvY3JsL3YyL3RjX2NsYXNzMV9MMV9DQV9WSS5jcmwwMwYDVR0lBCwwKgYIKwYBBQUHAwIG
CCsGAQUFBwMEBggrBgEFBQcDBwYKKwYBBAGCNxQCAjApBgNVHREEIjAggR5rYXkuZGllZGVy
aWNoc0B1bmkta29uc3RhbnouZGUwDQYJKoZIhvcNAQEFBQADgYEAcTvbHerOC4fWtku9Mb6r
z5DGROopvpCOtRS4QISajqmN6JCbjusSHTI4G8G5QLqBJB0Wg00p2v9nJasB9ZPz4h59Eyx8
FkQiyFt4jlo8JCHsCNSZ98bJsa73N0y3nOvQXChw8HLLhNq0r9hszPah9FcexLlXoUawmtaG
TYELBGcxggO0MIIDsAIBATCBjzB8MQswCQYDVQQGEwJERTEcMBoGA1UEChMTVEMgVHJ1c3RD
ZW50ZXIgR21iSDElMCMGA1UECxMcVEMgVHJ1c3RDZW50ZXIgQ2xhc3MgMSBMMSBDQTEoMCYG
A1UEAxMfVEMgVHJ1c3RDZW50ZXIgQ2xhc3MgMSBMMSBDQSBWSQIPAMYpAAEAAi4JCW7o2XnM
MAkGBSsOAwIaBQCgggH5MBgGCSqGSIb3DQEJAzELBgkqhkiG9w0BBwEwHAYJKoZIhvcNAQkF
MQ8XDTA5MDczMDE4NTYxNFowIwYJKoZIhvcNAQkEMRYEFFHzcLuH
+jTkW7daRwRBDYwU1P54
MFIGCSqGSIb3DQEJDzFFMEMwCgYIKoZIhvcNAwcwDgYIKoZIhvcNAwICAgCAMA0GCCqGSIb3
DQMCAgFAMAcGBSsOAwIHMA0GCCqGSIb3DQMCAgEoMIGgBgkrBgEEAYI3EAQxgZIwgY8wfDEL
MAkGA1UEBhMCREUxHDAaBgNVBAoTE1RDIFRydXN0Q2VudGVyIEdtYkgxJTAjBgNVBAsTHFRD
IFRydXN0Q2VudGVyIENsYXNzIDEgTDEgQ0ExKDAmBgNVBAMTH1RDIFRydXN0Q2VudGVyIENs
YXNzIDEgTDEgQ0EgVkkCDwDGKQABAAIuCQlu6Nl5zDCBogYLKoZIhvcNAQkQAgsxgZKggY8w
fDELMAkGA1UEBhMCREUxHDAaBgNVBAoTE1RDIFRydXN0Q2VudGVyIEdtYkgxJTAjBgNVBAsT
HFRDIFRydXN0Q2VudGVyIENsYXNzIDEgTDEgQ0ExKDAmBgNVBAMTH1RDIFRydXN0Q2VudGVy
IENsYXNzIDEgTDEgQ0EgVkkCDwDGKQABAAIuCQlu6Nl5zDANBgkqhkiG9w0BAQEFAASCAQAw
8wlcwpSWk0jXsjHcylqYlQvIrFcJ02zUaEjW+r0aHNmRmS62v/
ILa8BVMaWc7j4rInYjcRQ/
4MeBwqMOD7iK2usWIvp32SDnYdNY5qE4Jhn6F0eOLEpOZgET59XMYtzgBeMz2x/
fZUxDRxEo
0HeTHXjSv+kHeaPOVgkhoT5MbsV3Qvr7aBPW+yYO97Ek4FKxWX0AtgmqcWiK
+hGTaxX5vELw
ux4fjVWHKz66uovDStrp1Pgla0SIjbWyq7GYV0ol4Xq13r2IQgsR7SNV5hqFXSKT
+M0YI44y
I2Hfyc46XJ0pW9KFOO4DNxyLykju8ZdTC9OHSu503jsGiaJQTK/6AAAAAAAA
--------------ms000208020902090005010407--
------------------------------
Date: Thu, 30 Jul 2009 21:13:45 +0200
From: Luca Jovine <[email protected]>
Subject: Postdoctoral position in protein crystallography at
Karolinska Institutet
POSTDOCTORAL FELLOWSHIP IN PROTEIN CRYSTALLOGRAPHY AT KAROLINSKA
INSTITUT=
ET
As part of a collaboration between the laboratories of Rune
Toftg=E5rd an=
d Luca Jovine at the =
KI Department of Biosciences and Nutrition and the Center for
Biosciences=
=2C a postdoctoral =
position is immediately available on structural studies of key
players in=
the Hedgehog =
signaling pathway=2E
The Department of Biosciences and Nutrition has state-of-the-art
equipmen=
t for protein =
expression=2C purification and in house X-ray data collection=3B
frequent=
access to synchrotron =
sites and excellent computational facilities are also available=2E
With t=
heir close integration of =
academic=2C clinical and industrial research=2C KI and the Center
for Bio=
sciences offer a highly =
international and collaborative environment for biomedical studies=2E
Highly motivated candidates with experience in protein expression
(partic=
ularly in insect =
and/or mammalian cells)=2C purification=2C crystallization and
structure =
determination=2C are =
encouraged to apply by e-mailing a curriculum vitae=2C summary of
researc=
h experience and =
interests=2C and names and contact information for two references to
Luca=
Jovine =
(luca=2Ejovine=40ki=2Ese)=2E Please note that=2C in order to be
eligible =
for this scholarship=2C candidates =
should be non-Swedish citizens who have obtained a doctorate or the
equiv=
alent outside =
Sweden=2E Deadline for application is 1 October 2009=2E
------------------------------------------------
Luca Jovine=2C Ph=2ED=2E
Group Leader=2C Protein Crystallography Unit
Karolinska Institutet
Department of Biosciences and Nutrition
H=E4lsov=E4gen 7=2C SE-141 57 Huddinge=2C Sweden
Voice=3A +46=2E(0)8=2E6083-301 FAX=3A +46=2E(0)8=2E6089-290
E-mail=3A luca=2Ejovine=40ki=2Ese
W3=3A http=3A//jovinelab=2Eorg
------------------------------------------------
------------------------------
Date: Thu, 30 Jul 2009 12:29:09 -0700
From: Donnie Berkholz <[email protected]>
Subject: Re: question of extra high B factor
--/9DWx/yDrRhgMJTb
Content-Type: text/plain; charset=us-ascii
Content-Disposition: inline
Content-Transfer-Encoding: quoted-printable
On 00:00 Fri 31 Jul , Jiamu Du wrote:
Dear All,
I am refining a structure of a complex between of 50kD protein and
a 20kD
glycosylated protien. The data is of 2.9 A resolution. The wilson B
factor
is as high as 86.3 A^2.
The refinement seems well with R/Rf of 0.21/0.25. But the B-factor
is ext=
ra
high. For the 50kD part, the average B factor is 76.5 A^2. But the
B fact=
or
of the 20 kD glycosylated protein is as high as 133.3 A^2. Although
the
electron density looks fine, even the sugar chain is seen clearly.
My question is:
1. How to reduce the B factor to a reasonable level?
2. If it can not be redueced, when I published it, is this value
acceptab=
le?
3. In the same of similar resolutionIs, is there some other
structures li=
ke
this situation? A component or a subunit of the protein has a
extra high=
B
factor as high as 130.
Our group published a paper a few years ago with an average B of 80
A^2=20
(JMB 328:893 (2003)). One referee had some objections to this, so
we=20
performed analyses of the whole Protein Data Bank for proteins at=20
2.5-3.0 A (see Fig. 2D in the paper). You may find this figure
useful if=20
you need to convince anyone.
--=20
Thanks,
Donnie
Donnie Berkholz
P. Andrew Karplus lab
Oregon State University
--/9DWx/yDrRhgMJTb
Content-Type: application/pgp-signature
Content-Disposition: inline
-----BEGIN PGP SIGNATURE-----
Version: GnuPG v2.0.11 (GNU/Linux)
iEYEABECAAYFAkpx9IUACgkQXVaO67S1rtvQfgCbBMtImQXESgPibA1Jy3ud4YP8
oCwAn1UG7SWOlclYAVw3eNRL3LHTqTe/
=gKGd
-----END PGP SIGNATURE-----
--/9DWx/yDrRhgMJTb--
------------------------------
Date: Thu, 30 Jul 2009 17:53:15 -0400
From: Jim Fairman <[email protected]>
Subject: Re: OpenGL Stereo 3D on 120 Hz LCDs, at last!
--0015174be078b3c278046ff354f1
Content-Type: text/plain; charset=ISO-8859-1
Content-Transfer-Encoding: 7bit
Just got this working on a machine with a Quadro FX 3800. Stereo
looks
great on both Pymol (v1.1) and the latest build of WinCoot (v0.6 build
2172).
On Fri, Jul 17, 2009 at 1:20 AM, Warren DeLano <[email protected]>
wrote:
FYI, for folks not subscribed to pymol-users:
nVidia today released beta drivers which at last enable OpenGL-
based stereo
3D visualization on 120 Hz LCDs using Quadro graphics cards. So
long as you
are willing to put up with Windows, you can finally abandon those
old CRTs
without spending a fortune and without sacrificing quality of the
stereo 3D
effect.
Details posted at http://www.pymol.org
Cheers,
Warren
--
Jim Fairman
Graduate Research Assistant
Department of Biochemistry, Cellular, and Molecular Biology (BCMB)
University of Tennessee -- Knoxville
216-368-3337 [email protected] [email protected]
--0015174be078b3c278046ff354f1
Content-Type: text/html; charset=ISO-8859-1
Content-Transfer-Encoding: quoted-printable
Just got this working on a machine with a Quadro FX 3800.=A0 Stereo
looks g=
reat on both Pymol (v1.1) and the latest build of WinCoot (v0.6
build 2172)=
.<br><br><div class=3D"gmail_quote">On Fri, Jul 17, 2009 at 1:20 AM,
Warren=
DeLano <span dir=3D"ltr"><<a
href=3D"mailto:[email protected]";>war...@d=
elsci.com</a>></span> wrote:<br>
<blockquote class=3D"gmail_quote" style=3D"border-left: 1px solid
rgb(204, =
204, 204); margin: 0pt 0pt 0pt 0.8ex; padding-left: 1ex;">
<div link=3D"blue" vlink=3D"purple" lang=3D"EN-US">
<div>
<p><font size=3D"2" face=3D"Arial"><span style=3D"font-size: 10pt;
font-fam=
ily: Arial;">FYI, for folks not subscribed to pymol-users:=A0 </
span></font=
</p>
<p><font size=3D"2" face=3D"Arial"><span style=3D"font-size: 10pt;
font-fam=
ily: Arial;">=A0</span></font></p>
<p><font size=3D"2" face=3D"Arial"><span style=3D"font-size: 10pt;
font-fam=
ily: Arial;">nVidia today released beta drivers which at last enable
OpenGL=
-based
stereo 3D visualization on 120 Hz LCDs using Quadro graphics
cards.=A0 So l=
ong
as you are willing to put up with Windows, you can finally abandon
those ol=
d CRTs
without spending a fortune and without sacrificing quality of the
stereo 3D
effect.</span></font></p>
<p><font size=3D"2" face=3D"Arial"><span style=3D"font-size: 10pt;
font-fam=
ily: Arial;">=A0</span></font></p>
<p><font size=3D"2" face=3D"Arial"><span style=3D"font-size: 10pt;
font-fam=
ily: Arial;">Details posted at <a href=3D"http://www.pymol.org/";
target=3D"=
_blank">http://www.pymol.org</a></span></font></p>
<p><font size=3D"2" face=3D"Arial"><span style=3D"font-size: 10pt;
font-fam=
ily: Arial;">=A0</span></font></p>
<p><font size=3D"2" face=3D"Arial"><span style=3D"font-size: 10pt;
font-fam=
ily: Arial;">Cheers,</span></font></p>
<p><font size=3D"2" face=3D"Arial"><span style=3D"font-size: 10pt;
font-fam=
ily: Arial;">Warren</span></font><font size=3D"2"
face=3D"Arial"><span styl=
e=3D"font-size: 10pt; font-family: Arial;"></span></font></p>
<p><font size=3D"2" face=3D"Arial"><span style=3D"font-size: 10pt;
font-fam=
ily: Arial;">=A0</span></font></p>
</div>
</div>
</blockquote></div><br><br clear=3D"all"><br>-- <br>Jim
Fairman<br>Graduate=
Research Assistant<br>Department of Biochemistry, Cellular, and
Molecular =
Biology (BCMB)<br>University of Tennessee --
Knoxville<br>216-368-3337 <a h=
ref=3D"mailto:[email protected]";>[email protected]</a> <a href=3D"mailto:jame=
[email protected]">[email protected]</a><br>
--0015174be078b3c278046ff354f1--
------------------------------
Date: Thu, 30 Jul 2009 15:23:04 -0700
From: James Holton <[email protected]>
Subject: Re: Foils for energy calibration
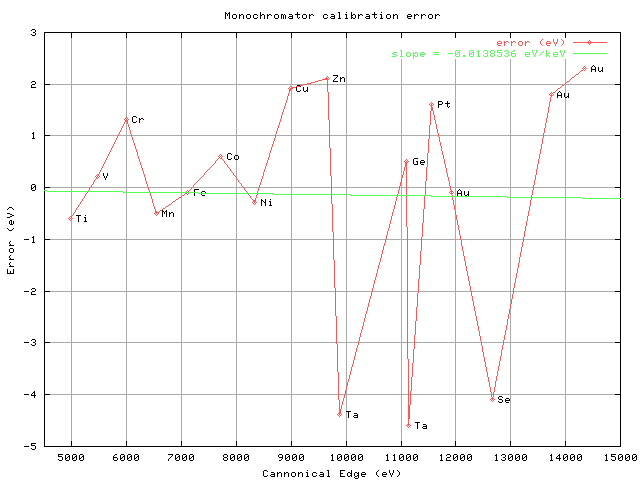
At ALS, we have a box of foils from EXAFS Materials that seems to
get=20
passed around from beamline to beamline. I ran absorption scans on
17=20
edges from the metals in the box one day, and found that there was=20
considerable scatter in the expected vs observed edge positions:
http://bl831.als.lbl.gov/~jamesh/pickup/mono_calib.png
Here I have plotted the "correct" position of each edge as
determined by=20
Bearden & Burr (1967), against the edge I determined using the
criterion=20
recommended in the "Reference Spectra" document in the=20
exafsmaterials.com website: the first inflection point in the
derivative=20
spectrum. I think a large amount of the scatter is because my mono=20
(like many PX/MX beamlines) is Si(111) and not Si(220) like the one
used=20
to determine the reference spectra. It is not hard to imagine how=20
blurring the spectrum with a wider energy spread of the incident
beam=20
will shift the position of the "edge". One could try to use the=20
electron binding energy tabulated in the "little orange book":
http://xdb.lbl.gov/Section1/Sec_1-1.html
but these do not always take into account the "near edge" features
(like=20
the white line from SeMet) which change depending on the chemical=20
environment around the metal, radiation damage, etc. It would be
nice=20
if someone could calibrate some standard reference materials using
a=20
Si(111) monochromator, but I don't know of anyone who has done this.
However, another way to get your x-ray wavelength is using Bragg's
law:
lambda =3D 2*d*sin(theta)
and the d-spacing of silicon is known to be 5.43159 =B1 0.00020 A,
and=20
NIST will sell you certified Si powder:
https://www-s.nist.gov/srmors/view_detail.cfm?srm=3D640d
The problem here is that although you know d very accurately, the
error=20
in lambda is dominated by sin(theta), or rather the uncertainty in
your=20
detector distance. The pixel field on most detectors is actually
quite=20
accurate, as a NIST-traceable calibration is used to make the
pinhole=20
calibration mask, and the encoder on most detector distance stages
is=20
very accurate for relative moves (counting ticks on the encoder).
But=20
there is always an offset from the "zero" position predicted by the=20
encoder to the true center of rotation that is hard to know.=20
Nevertheless, all you really want is for the d-spacing of silicon
powder=20
rings to be right at all detector distances. You can use the
program=20
FIT2D to refine the wavelength, distance, detector tilt, etc. or
any=20
combination thereof for a given image, but you will find that the=20
repeatability of such a fit (using different starting parameters) is
not=20
great because the distance and wavelength are highly correlated. =20
However, there is a way around this:
Since we know that a relative move of the distance will be
accurate,=20
there should be one and only one offset that you can add to the
recorded=20
value of distance of each image to make it the "right" distance.
You=20
can define the "right" offset as the one where FIXing the resulting=20
"right" distance in FIT2D and refining everything else gives you
the=20
same refined value for the wavelength from every image. You need
to=20
manually "refine" this offset for a few rounds. What you will
generally=20
see is that the graph of fitted wavelength vs the distance is a
straight=20
line, and you want to make the slope of this line to be zero. =20
Eventually, you will arrive at some offset that gives you the
smallest=20
spread in refined wavelength values. The average refined wavelength
is=20
then the "true" wavelength. Should be able to get it within one or
two=20
eV. Perhaps more if you take a lot of silicon powder images.
At ALS beamlines 8.3.1 and 12.3.1 I have done both kinds of
calibration,=20
and I am fairly certain I get the wavelength accurate to within 1 eV
by=20
calibrating the half-way-up point of an absorption scan of a ~122
micron=20
thick copper metal foil to 8979.0 eV.
-James Holton
MAD Scientist
Richard Gillilan wrote:
In the past we've used elemental foils from exafsmaterials.com for=20
energy calibration of our MAD beamline. These standards are for
EXAFS=20
and XANES. Most are thin (5 micron) metal foils.
Has anyone had experience with other sources of standards or other=20
forms (such as compounds rather than pure elements)?
I notice that a number of companies offer XRF standard kits.
Richard Gillilan
MacCHESS
------------------------------
End of CCP4BB Digest - 29 Jul 2009 to 30 Jul 2009 (#2009-207)
*************************************************************
{kind=link}