Alex, With regard to scalepack, be sure you use NO MERGE ORIGINAL INDEX, not just NO MERGE.
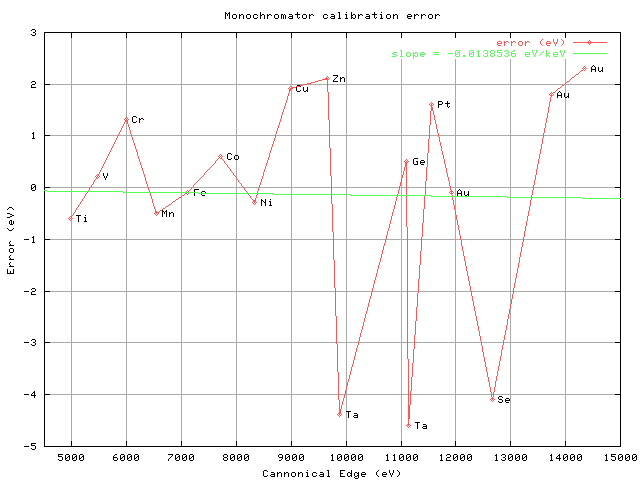
Marian Szebenyi MacCHESS > Dear all: > > I am trying to solve a structure from apparently a hexagonal crystal. > I indexed and scaled data in P6 in Scalepack (with merging) then used > Scalepack2mtz (with ensure unique reflections and add Rfree as well as > the truncate procedure), and then attempted to run molecular > replacement with Phaser. Now the problem appeared - Phaser immediately > quits with the following error message "FATAL RUNTIME ERROR: > Reflections are not a unique set by symmetry". I do not understand > this at all. > > I also tried running scalepack using the NO MERGE macro as people have > indicated earlier on this bb (thank you again!, I also checked the > scl.in that is written out and it had the NO MERGE statement), and > then tried to run pointless to verify the spacegroup but the program > complained the reflections are merged (that is impossible, I checked > the number of reflections in the unmerged and merged files and they > were different as one would expect). I repeated the procedures several > times and I always get the same errors. I can't make any sense of this > and I can't move forward. Any ideas? > > Many thanks, > Alex > > > > > > > On Jul 30, 2009, at 7:03 PM, CCP4BB automatic digest system wrote: > >> There are 13 messages totalling 1026 lines in this issue. >> >> Topics of the day: >> >> 1. refmac failed message (2) >> 2. question of extra high B factor (6) >> 3. Coot:findwaters in REFMAC (2) >> 4. Postdoctoral position in protein crystallography at Karolinska >> Institutet >> 5. OpenGL Stereo 3D on 120 Hz LCDs, at last! >> 6. Foils for energy calibration >> >> ---------------------------------------------------------------------- >> >> Date: Thu, 30 Jul 2009 10:50:13 GMT >> From: Elad Binshtien <[email protected]> >> Subject: refmac failed message >> >> This is a multi-part message in MIME format. >> >> >> ----8d5c55134e491a1d1bdd >> Content-Type: text/plain; charset=utf-8 >> Content-Disposition: inline >> Content-Transfer-Encoding: quoted-printable >> >> Dear all=2C >> >> >> I am refining a structure in Refmac at 2=2E2 A in win OS=2E=C2=A0 >> Howeve= >> r=2C >> Refmac failed and send this message=3A =C2=A0 =C2=A0 =C2=A0=C2=A0 >> forrtl= >> =3A error (72)=3A floating overflow=C2=A0 = >> >> >> Thank you in advance for your any helpful suggestions=2E=C2=A0 = >> >> >> >> Best=2C >> Elad >> >> >> Elad Binshtein >> Ph=2ED student = >> >> Department of Life Science = >> >> Ben Gurion University of the Negev >> Ph=3A 972-8-6461325=E2=80=8E >> >> ----8d5c55134e491a1d1bdd >> Content-Type: text/html; charset=utf-8 >> Content-Disposition: inline >> Content-Transfer-Encoding: quoted-printable >> >> Dear all=2C=3Cbr=3E=3Cbr=3E=3Cbr=3EI am refining a structure in >> Refmac a= >> t 2=2E2 A in win OS=2E=26nbsp=3B However=2C >> Refmac failed and send this message=3A =26nbsp=3B =26nbsp=3B >> =26nbsp=3B=26= >> nbsp=3B forrtl=3A error (72)=3A floating overflow=26nbsp=3B >> =3Cbr=3E=3Cb= >> r=3E Thank you in advance for your any helpful >> suggestions=2E=26nbsp=3B = >> = >> 3Cbr >> =3E=3Cbr=3E=3Cbr=3EBest=2C=3Cbr=3EElad=3Cbr=3E=3CBR=3E=3CBR=3EElad = >> Binshtein=3Cbr=3EPh=2ED student =3Cbr=3EDepartment of Life Science >> =3Cbr= >> =3EBen Gurion University of the Negev=3Cbr=3EPh=3A 972-8-6461325=3C/ >> BR=3E= >> =3C/BR=3E=E2=80=8E >> >> ----8d5c55134e491a1d1bdd-- >> >> ------------------------------ >> >> Date: Thu, 30 Jul 2009 13:25:43 +0100 >> From: "Stein, ND (Norman)" <[email protected]> >> Subject: Re: refmac failed message >> >> This is a multi-part message in MIME format. >> >> ------_=_NextPart_001_01CA1110.DB7CC0CB >> Content-Type: text/plain; >> charset="windows-1255" >> Content-Transfer-Encoding: quoted-printable >> >> Hi Elad >> =20 >> You don't say which version of Refmac you are using but I think the = >> first thing to do would be to try the latest version (5.5.0102). You >> can = >> pick this up from >> =20 >> ftp://ftp.ccp4.ac.uk/nds/windows/refmac_5.5.0102/refmac5.exe >> =20 >> Copy this exe file to the bin subdirectory of your CCP4 >> installation. = >> (Probably wise to make a backup copy of the existing refmac5.exe >> first). >> =20 >> Best wishes >> =20 >> Norman Stein >> CCP4 >> >> ________________________________ >> >> From: CCP4 bulletin board [mailto:[email protected]] On Behalf >> Of = >> Elad Binshtien >> Sent: 30 July 2009 11:50 >> To: [email protected] >> Subject: [ccp4bb] refmac failed message >> >> >> Dear all, >> >> >> I am refining a structure in Refmac at 2.2 A in win OS. However, >> Refmac = >> failed and send this message: forrtl: error (72): floating = >> overflow =20 >> >> Thank you in advance for your any helpful suggestions. =20 >> >> >> Best, >> Elad >> >> >> Elad Binshtein >> Ph.D student=20 >> Department of Life Science=20 >> Ben Gurion University of the Negev >> Ph: 972-8-6461325 >> >> =FD=20 >> >> -- =0AScanned by iCritical.=0A >> >> ------_=_NextPart_001_01CA1110.DB7CC0CB >> Content-Type: text/html; >> charset="windows-1255" >> Content-Transfer-Encoding: quoted-printable >> >> <!DOCTYPE HTML PUBLIC "-//W3C//DTD HTML 4.0 Transitional//EN"> >> <HTML><HEAD> >> <META http-equiv=3DContent-Type content=3D"text/html; = >> charset=3Dwindows-1255"> >> <META content=3D"MSHTML 6.00.2900.3562" name=3DGENERATOR></HEAD> >> <BODY> >> <DIV dir=3Dltr align=3Dleft><SPAN class=3D419481712-30072009><FONT = >> face=3DArial=20 >> color=3D#0000ff size=3D2>Hi Elad</FONT></SPAN></DIV> >> <DIV dir=3Dltr align=3Dleft><SPAN class=3D419481712-30072009><FONT = >> face=3DArial=20 >> color=3D#0000ff size=3D2></FONT></SPAN> </DIV> >> <DIV dir=3Dltr align=3Dleft><SPAN class=3D419481712-30072009><FONT = >> face=3DArial=20 >> color=3D#0000ff size=3D2>You don't say which version of Refmac you >> are = >> using but I=20 >> think the first thing to do would be to try the latest = >> version (5.5.0102).=20 >> You can pick this up from</FONT></SPAN></DIV> >> <DIV dir=3Dltr align=3Dleft><SPAN class=3D419481712-30072009><FONT = >> face=3DArial=20 >> color=3D#0000ff size=3D2></FONT></SPAN> </DIV> >> <DIV dir=3Dltr align=3Dleft><SPAN class=3D419481712-30072009><FONT = >> face=3DArial=20 >> color=3D#0000ff size=3D2><A=20 >> href=3D"ftp://ftp.ccp4.ac.uk/nds/windows/refmac_5.5.0102/ >> refmac5.exe">ftp= >> ://ftp.ccp4.ac.uk/nds/windows/refmac_5.5.0102/refmac5.exe</A></ >> FONT></SPA= >> N></DIV> >> <DIV dir=3Dltr align=3Dleft><SPAN class=3D419481712-30072009><FONT = >> face=3DArial=20 >> color=3D#0000ff size=3D2></FONT></SPAN> </DIV> >> <DIV dir=3Dltr align=3Dleft><SPAN class=3D419481712-30072009><FONT = >> face=3DArial=20 >> color=3D#0000ff size=3D2>Copy this exe file to the bin subdirectory >> of=20 >> your CCP4 installation. (Probably wise to make a backup >> copy = >> of the=20 >> existing refmac5.exe first).</FONT></SPAN></DIV> >> <DIV dir=3Dltr align=3Dleft><SPAN class=3D419481712-30072009><FONT = >> face=3DArial=20 >> color=3D#0000ff size=3D2></FONT></SPAN> </DIV> >> <DIV dir=3Dltr align=3Dleft><SPAN class=3D419481712-30072009><FONT = >> face=3DArial=20 >> color=3D#0000ff size=3D2>Best wishes</FONT></SPAN></DIV> >> <DIV dir=3Dltr align=3Dleft><SPAN class=3D419481712-30072009><FONT = >> face=3DArial=20 >> color=3D#0000ff size=3D2></FONT></SPAN> </DIV> >> <DIV dir=3Dltr align=3Dleft><SPAN class=3D419481712-30072009><FONT = >> face=3DArial=20 >> color=3D#0000ff size=3D2>Norman Stein</FONT></SPAN></DIV> >> <DIV dir=3Dltr align=3Dleft><SPAN class=3D419481712-30072009><FONT = >> face=3DArial=20 >> color=3D#0000ff size=3D2>CCP4</FONT></SPAN></DIV><BR> >> <DIV class=3DOutlookMessageHeader lang=3Den-us dir=3Dltr align=3Dleft> >> <HR tabIndex=3D-1> >> <FONT face=3DTahoma size=3D2><B>From:</B> CCP4 bulletin board=20 >> [mailto:[email protected]] <B>On Behalf Of </B>Elad=20 >> Binshtien<BR><B>Sent:</B> 30 July 2009 11:50<BR><B>To:</B>=20 >> [email protected]<BR><B>Subject:</B> [ccp4bb] refmac failed=20 >> message<BR></FONT><BR></DIV> >> <DIV></DIV>Dear all,<BR><BR><BR>I am refining a structure in Refmac >> at = >> 2.2 A in=20 >> win OS. However, Refmac failed and send this message: = >> =20 >> forrtl: error (72): floating overflow >> <BR><BR>Thank = >> you in=20 >> advance for your any helpful suggestions. =20 >> <BR><BR><BR>Best,<BR>Elad<BR><BR><BR>Elad Binshtein<BR>Ph.D student=20 >> <BR>Department of Life Science <BR>Ben Gurion University of the = >> Negev<BR>Ph:=20 >> 972-8-6461325<BR><BR>=FD >> <br>= >> <p>-- =0A<BR>Scanned by iCritical.=0A</p> >> <br>= >> </BODY></HTML> >> >> ------_=_NextPart_001_01CA1110.DB7CC0CB-- >> >> ------------------------------ >> >> Date: Fri, 31 Jul 2009 00:00:25 +0800 >> From: Jiamu Du <[email protected]> >> Subject: question of extra high B factor >> >> --00221504874fd6c69c046fee6655 >> Content-Type: text/plain; charset=ISO-8859-1 >> Content-Transfer-Encoding: 7bit >> >> Dear All, >> I am refining a structure of a complex between of 50kD protein and a >> 20kD >> glycosylated protien. The data is of 2.9 A resolution. The wilson B >> factor >> is as high as 86.3 A^2. >> The refinement seems well with R/Rf of 0.21/0.25. But the B-factor >> is extra >> high. For the 50kD part, the average B factor is 76.5 A^2. But the B >> factor >> of the 20 kD glycosylated protein is as high as 133.3 A^2. Although >> the >> electron density looks fine, even the sugar chain is seen clearly. >> My question is: >> 1. How to reduce the B factor to a reasonable level? >> 2. If it can not be redueced, when I published it, is this value >> acceptable? >> 3. In the same of similar resolutionIs, is there some other >> structures like >> this situation? A component or a subunit of the protein has a extra >> high B >> factor as high as 130. >> >> Thanks and best wishes. >> >> >> -- >> Jiamu Du, Ph.D. >> State Key Laboratory of Molecular Biology >> Institute of Biochemistry and Cell Biology >> Shanghai Institutes for Biological Sciences >> Chinese Academy of Sciences >> 320 Yue-Yang Road >> Shanghai 200031 >> P. R. China >> Tel: +86-21-5492-1117 >> E-mail: [email protected] >> >> --00221504874fd6c69c046fee6655 >> Content-Type: text/html; charset=ISO-8859-1 >> Content-Transfer-Encoding: quoted-printable >> >> Dear All,<br>I am refining a structure of a complex between of 50kD >> protein= >> and a 20kD glycosylated protien. The data is of 2.9 A resolution. >> The wils= >> on B factor is as high as 86.3 A^2.<br>The refinement seems well >> with R/Rf = >> of 0.21/0.25. But the B-factor is extra high. For the 50kD part, the >> averag= >> e B factor is 76.5 A^2. But the B factor of the 20 kD glycosylated >> protein = >> is as high as 133.3 A^2. Although the electron density looks fine, >> even the= >> sugar chain is seen clearly.<br> >> My question is:<br>1. How to reduce the B factor to a reasonable >> level?<br>= >> 2. If it can not be redueced, when I published it, is this value >> acceptable= >> ?<br>3. In the same of similar resolutionIs, is there some other >> structures= >> like this situation?=A0 A component or a subunit of the protein has >> a extr= >> a high B factor as high as 130.<br> >> <br>Thanks and best wishes.<br><br clear=3D"all"><br>-- <br>Jiamu >> Du, Ph.D.= >> <br>State Key Laboratory of Molecular Biology<br>Institute of >> Biochemistry = >> and Cell Biology<br>Shanghai Institutes for Biological >> Sciences<br>Chinese = >> Academy of Sciences<br> >> 320 Yue-Yang Road<br>Shanghai 200031<br>P. R. China<br>Tel: >> +86-21-5492-111= >> 7<br>E-mail: <a href=3D"mailto:[email protected]";>[email protected]</ >> a><br> >> >> --00221504874fd6c69c046fee6655-- >> >> ------------------------------ >> >> Date: Thu, 30 Jul 2009 09:32:26 -0700 >> From: Pavel Afonine <[email protected]> >> Subject: Re: question of extra high B factor >> >> Hi Jiamu, >> >>> My question is: >>> 1. How to reduce the B factor to a reasonable level? >>> 3. In the same of similar resolutionIs, is there some other >>> structures >>> like this situation? >> >> POLYGON tool is (one of) your friend(s) to answer this question (apart >> from debatable one about "a reasonable level"): >> >> Acta Cryst. D65, 297-300 (2009). >> >> Now it is available in PHENIX (from the latest nightly builds): >> http://www.phenix-online.org/ >> >> Please let me know if you have any questions about it. >> >> Pavel. >> >> ------------------------------ >> >> Date: Thu, 30 Jul 2009 17:47:55 +0100 >> From: Clemens Vonrhein <[email protected]> >> Subject: Re: question of extra high B factor >> >> Dear Jiamu, >> >> On Fri, Jul 31, 2009 at 12:00:25AM +0800, Jiamu Du wrote: >>> I am refining a structure of a complex between of 50kD protein and >>> a 20kD >>> glycosylated protien. The data is of 2.9 A resolution. The wilson B >>> factor >>> is as high as 86.3 A^2. >>> The refinement seems well with R/Rf of 0.21/0.25. But the B-factor >>> is extra >>> high. For the 50kD part, the average B factor is 76.5 A^2. But the >>> B factor >>> of the 20 kD glycosylated protein is as high as 133.3 A^2. Although >>> the >>> electron density looks fine, even the sugar chain is seen clearly. >>> My question is: >>> 1. How to reduce the B factor to a reasonable level? >> >> How do you define 'reasonable'? And why would you want to reduce this >> anyway? >> >> ( 50*76.5 + 20*133.3 ) / 70 = 92.7 >> >> which seems fairly close to the Wilson B of 86.3, right? >> >>> 2. If it can not be redueced, when I published it, is this value >>> acceptable? >> >> Better question: is it the right value? Remember it is >> >> results -> publish >> >> and not >> >> publish <- results >> >> ;-) >> >> If they're correct than they are acceptable (I would accept those >> values). >> >>> 3. In the same of similar resolutionIs, is there some other >>> structures like >>> this situation? A component or a subunit of the protein has a >>> extra high B >>> factor as high as 130. >> >> I'm sure hundreds of them ... but I'm sure you want your structure to >> stand on its own, so don't look too close at other structures and >> repeat the various mistakes we've all made and that are now set in >> stone in some old PDB file. >> >> Cheers >> >> Clemens >> >> -- >> >> *************************************************************** >> * Clemens Vonrhein, Ph.D. vonrhein AT GlobalPhasing DOT com >> * >> * Global Phasing Ltd. >> * Sheraton House, Castle Park >> * Cambridge CB3 0AX, UK >> *-------------------------------------------------------------- >> * BUSTER Development Group (http://www.globalphasing.com) >> *************************************************************** >> >> ------------------------------ >> >> Date: Fri, 31 Jul 2009 00:02:42 +0800 >> From: Jiamu Du <[email protected]> >> Subject: Re: question of extra high B factor >> >> --0022152d604d09ee86046fee6f07 >> Content-Type: text/plain; charset=ISO-8859-1 >> Content-Transfer-Encoding: 7bit >> >> I am using TLS refinement with Phenix for the structure refinement. >> >> >> On Fri, Jul 31, 2009 at 12:00 AM, Jiamu Du <[email protected]> wrote: >> >>> Dear All, >>> I am refining a structure of a complex between of 50kD protein and >>> a 20kD >>> glycosylated protien. The data is of 2.9 A resolution. The wilson B >>> factor >>> is as high as 86.3 A^2. >>> The refinement seems well with R/Rf of 0.21/0.25. But the B-factor >>> is extra >>> high. For the 50kD part, the average B factor is 76.5 A^2. But the >>> B factor >>> of the 20 kD glycosylated protein is as high as 133.3 A^2. Although >>> the >>> electron density looks fine, even the sugar chain is seen clearly. >>> My question is: >>> 1. How to reduce the B factor to a reasonable level? >>> 2. If it can not be redueced, when I published it, is this value >>> acceptable? >>> 3. In the same of similar resolutionIs, is there some other >>> structures like >>> this situation? A component or a subunit of the protein has a >>> extra high B >>> factor as high as 130. >>> >>> Thanks and best wishes. >>> >>> >>> -- >>> Jiamu Du, Ph.D. >>> State Key Laboratory of Molecular Biology >>> Institute of Biochemistry and Cell Biology >>> Shanghai Institutes for Biological Sciences >>> Chinese Academy of Sciences >>> 320 Yue-Yang Road >>> Shanghai 200031 >>> P. R. China >>> Tel: +86-21-5492-1117 >>> E-mail: [email protected] >>> >> >> >> >> -- >> Jiamu Du, Ph.D. >> State Key Laboratory of Molecular Biology >> Institute of Biochemistry and Cell Biology >> Shanghai Institutes for Biological Sciences >> Chinese Academy of Sciences >> 320 Yue-Yang Road >> Shanghai 200031 >> P. R. China >> Tel: +86-21-5492-1117 >> E-mail: [email protected] >> >> --0022152d604d09ee86046fee6f07 >> Content-Type: text/html; charset=ISO-8859-1 >> Content-Transfer-Encoding: quoted-printable >> >> I am using TLS refinement with Phenix for the structure >> refinement.<br><br>= >> <br><div class=3D"gmail_quote">On Fri, Jul 31, 2009 at 12:00 AM, >> Jiamu Du <= >> span dir=3D"ltr"><<a >> href=3D"mailto:[email protected]";>[email protected] >> = >> </a>></span> wrote:<br> >> <blockquote class=3D"gmail_quote" style=3D"border-left: 1px solid >> rgb(204, = >> 204, 204); margin: 0pt 0pt 0pt 0.8ex; padding-left: 1ex;">Dear >> All,<br>I am= >> refining a structure of a complex between of 50kD protein and a 20kD >> glyco= >> sylated protien. The data is of 2.9 A resolution. The wilson B >> factor is as= >> high as 86.3 A^2.<br> >> The refinement seems well with R/Rf of 0.21/0.25. But the B-factor >> is extra= >> high. For the 50kD part, the average B factor is 76.5 A^2. But the B >> facto= >> r of the 20 kD glycosylated protein is as high as 133.3 A^2. >> Although the e= >> lectron density looks fine, even the sugar chain is seen clearly.<br> >> >> My question is:<br>1. How to reduce the B factor to a reasonable >> level?<br>= >> 2. If it can not be redueced, when I published it, is this value >> acceptable= >> ?<br>3. In the same of similar resolutionIs, is there some other >> structures= >> like this situation?=A0 A component or a subunit of the protein has >> a extr= >> a high B factor as high as 130.<br> >> >> <br>Thanks and best wishes.<br><br clear=3D"all"><br>-- <br>Jiamu >> Du, Ph.D.= >> <br>State Key Laboratory of Molecular Biology<br>Institute of >> Biochemistry = >> and Cell Biology<br>Shanghai Institutes for Biological >> Sciences<br>Chinese = >> Academy of Sciences<br> >> >> 320 Yue-Yang Road<br>Shanghai 200031<br>P. R. China<br>Tel: >> +86-21-5492-111= >> 7<br>E-mail: <a href=3D"mailto:[email protected]"; >> target=3D"_blank">jiamudu= >> @gmail.com</a><br> >> </blockquote></div><br><br clear=3D"all"><br>-- <br>Jiamu Du, >> Ph.D.<br>Stat= >> e Key Laboratory of Molecular Biology<br>Institute of Biochemistry >> and Cell= >> Biology<br>Shanghai Institutes for Biological Sciences<br>Chinese >> Academy = >> of Sciences<br> >> 320 Yue-Yang Road<br>Shanghai 200031<br>P. R. China<br>Tel: >> +86-21-5492-111= >> 7<br>E-mail: <a href=3D"mailto:[email protected]";>[email protected]</ >> a><br> >> >> --0022152d604d09ee86046fee6f07-- >> >> ------------------------------ >> >> Date: Thu, 30 Jul 2009 10:24:25 -0700 >> From: Huiying Li <[email protected]> >> Subject: Coot:findwaters in REFMAC >> >> I used Coot:findwaters facility in REFMAC (CCP4 6.1.1) to add waters >> to >> the refined structure. Often for the well ordered water sites the >> routine added two water molecules in single water site, with their >> distance (~1 Angs) way shorter than allowed hydrogen bonding >> distance. I >> have to remove the extra water molecules manually. >> >> Is this a intended feature? It seems to only create extra work to the >> user. What is the purpose of having different chain IDs for waters >> output >> from this routine? >> >> >> Huiying Li, Ph. D >> Department of Molecular Biology and Biochemistry >> Natural Sciences I, Rm 2443 >> University of California at Irvine >> Irvine, CA 92697, USA >> Tel: 949-824-4322(or -1953); Fax: 949-824-3280 >> email: [email protected] >> >> ------------------------------ >> >> Date: Thu, 30 Jul 2009 18:33:01 +0100 >> From: Paul Emsley <[email protected]> >> Subject: Re: Coot:findwaters in REFMAC >> >> Huiying Li wrote: >>> I used Coot:findwaters facility in REFMAC (CCP4 6.1.1) to add waters >>> to the refined structure. Often for the well ordered water sites the >>> routine added two water molecules in single water site, with their >>> distance (~1 Angs) way shorter than allowed hydrogen bonding >>> distance. >>> I have to remove the extra water molecules manually. >> >> Sounds like an old version. Things have improved. >> >>> >>> Is this a intended feature? It seems to only create extra work to the >>> user. >> >> :-( >> >>> What is the purpose of having different chain IDs for waters output >>> from this routine? >>> >> >> I thought (at the time) that it was sensible to add waters to a >> different chain ID to the protein atoms (I still do, in fact). There >> are others (somewhat less involved in model-building I suspect) that >> think otherwise. >> >> Paul. >> >> ------------------------------ >> >> Date: Thu, 30 Jul 2009 20:56:14 +0200 >> From: Kay Diederichs <[email protected]> >> Subject: Re: question of extra high B factor >> >> This is a cryptographically signed message in MIME format. >> >> --------------ms000208020902090005010407 >> Content-Type: text/plain; charset=ISO-8859-1; format=flowed >> Content-Transfer-Encoding: 7bit >> >> Jiamu Du schrieb: >>> Dear All, >>> I am refining a structure of a complex between of 50kD protein and a >>> 20kD glycosylated protien. The data is of 2.9 A resolution. The >>> wilson B >>> factor is as high as 86.3 A^2. >>> The refinement seems well with R/Rf of 0.21/0.25. But the B-factor is >>> extra high. For the 50kD part, the average B factor is 76.5 A^2. >>> But the >>> B factor of the 20 kD glycosylated protein is as high as 133.3 A^2. >>> Although the electron density looks fine, even the sugar chain is >>> seen >>> clearly. >>> My question is: >>> 1. How to reduce the B factor to a reasonable level? >> >> Please check out >> <http://strucbio.biologie.uni-konstanz.de/ccp4wiki/index.php/Refinement#help.2C_my_protein_has_high_B-factors.21 >> > >> >>> 2. If it can not be redueced, when I published it, is this value >>> acceptable? >> >> As there's nothing wrong with it, it can be published. >> >>> 3. In the same of similar resolutionIs, is there some other >>> structures >>> like this situation? A component or a subunit of the protein has a >>> extra high B factor as high as 130. >> >> Just look at the B-factors of new PDB entries which have a a >> resolution >> worse than 3 A (e.g., membrane proteins) - there are quite a few of >> those. >> >> HTH, >> >> Kay >> -- >> Kay Diederichs http://strucbio.biologie.uni- >> konstanz.de >> email: [email protected] Tel +49 7531 88 4049 Fax >> 3183 >> Fachbereich Biologie, Universitaet Konstanz, Box M647, D-78457 >> Konstanz >> >> --------------ms000208020902090005010407 >> Content-Type: application/x-pkcs7-signature; name="smime.p7s" >> Content-Transfer-Encoding: base64 >> Content-Disposition: attachment; filename="smime.p7s" >> Content-Description: S/MIME Cryptographic Signature >> >> MIAGCSqGSIb3DQEHAqCAMIACAQExCzAJBgUrDgMCGgUAMIAGCSqGSIb3DQEHAQAAoIIOlTCC >> BM8wggQ4oAMCAQICDwCUsAABAAJijbUfbuxyZDANBgkqhkiG9w0BAQUFADCBvDELMAkGA1UE >> BhMCREUxEDAOBgNVBAgTB0hhbWJ1cmcxEDAOBgNVBAcTB0hhbWJ1cmcxOjA4BgNVBAoTMVRD >> IFRydXN0Q2VudGVyIGZvciBTZWN1cml0eSBpbiBEYXRhIE5ldHdvcmtzIEdtYkgxIjAgBgNV >> BAsTGVRDIFRydXN0Q2VudGVyIENsYXNzIDEgQ0ExKTAnBgkqhkiG9w0BCQEWGmNlcnRpZmlj >> YXRlQHRydXN0Y2VudGVyLmRlMB4XDTA4MDcxODExMzg1NFoXDTEwMTIzMTIyNTk1OVowfDEL >> MAkGA1UEBhMCREUxHDAaBgNVBAoTE1RDIFRydXN0Q2VudGVyIEdtYkgxJTAjBgNVBAsTHFRD >> IFRydXN0Q2VudGVyIENsYXNzIDEgTDEgQ0ExKDAmBgNVBAMTH1RDIFRydXN0Q2VudGVyIENs >> YXNzIDEgTDEgQ0EgVkkwgZ8wDQYJKoZIhvcNAQEBBQADgY0AMIGJAoGBAJQFP39OlsR >> +FrO6 >> sIC5tx+k3aTjsL4oCLrsrd2Z9nVMzrHxnvShlmxLhgElFThIoRzo7gr1mZbx2/ >> Gu3X51SEBN >> TkP3bV2U0jn9yxGo97oYWA/+cRUKEEsxfvbPX5DL992EupcEptQAVutm32so3dGi >> +nqe2mFX >> wx8wpDAnCkDRAgMBAAGjggIQMIICDDCBjgYIKwYBBQUHAQEEgYEwfzBMBggrBgEFBQcwAoZA >> aHR0cDovL3d3dy50cnVzdGNlbnRlci5kZS9jZXJ0c2VydmljZXMvY2FjZXJ0cy90Y19jbGFz >> c18xX2NhLmNydDAvBggrBgEFBQcwAYYjaHR0cDovL29jc3AudGNjbGFzczEudHJ1c3RjZW50 >> ZXIuZGUwDwYDVR0TAQH/BAUwAwEB/ >> zBKBgNVHSAEQzBBMD8GCSqCFAAsAQEBATAyMDAGCCsG >> AQUFBwIBFiRodHRwOi8vd3d3LnRydXN0Y2VudGVyLmRlL2d1aWRlbGluZXMwDgYDVR0PAQH >> / >> BAQDAgGGMB0GA1UdDgQWBBROm/Wy+91qa9xKB6/Ju2T/ >> FksmnzCB7AYDVR0fBIHkMIHhMIHe >> oIHboIHYhjtodHRwOi8vY3JsLnRjY2xhc3MxLnRydXN0Y2VudGVyLmRlL2NybC92Mi90Y19j >> bGFzc18xX2NhLmNybIaBmGxkYXA6Ly93d3cudHJ1c3RjZW50ZXIuZGUvQ049VEMlMjBUcnVz >> dENlbnRlciUyMENsYXNzJTIwMSUyMENBLE89VEMlMjBUcnVzdENlbnRlciUyMEFHLG91PXJv >> b3RjZXJ0cyxkYz10cnVzdGNlbnRlcixkYz1kZT9jZXJ0aWZpY2F0ZVJldm9jYXRpb25MaXN0 >> P2Jhc2U/MA0GCSqGSIb3DQEBBQUAA4GBAG4ApLln3RLqDiymjdk8SFs8S6acQryrkZB/ >> DJ2Q >> H6n+6LYqEjirh+/Zj8uweQL6p6d9g/dl4S1wpIGCVC33AdcM8qer/ >> Cn0qcQ6bdduesfTcdQv >> 5uZlSsyEdKNa44DVtOh3IkTLtFdcDiY4tQFriM7kVC5Xuhyfxo58V1vOM8r >> +MIIE3TCCBEag >> AwIBAgIPAMYpAAEAAi4JCW7o2XnMMA0GCSqGSIb3DQEBBQUAMHwxCzAJBgNVBAYTAkRFMRww >> GgYDVQQKExNUQyBUcnVzdENlbnRlciBHbWJIMSUwIwYDVQQLExxUQyBUcnVzdENlbnRlciBD >> bGFzcyAxIEwxIENBMSgwJgYDVQQDEx9UQyBUcnVzdENlbnRlciBDbGFzcyAxIEwxIENBIFZJ >> MB4XDTA5MDYxMTA3MzM1NVoXDTEwMDYxMjA3MzM1NVowWTELMAkGA1UEBhMCREUxGzAZBgNV >> BAMTEkRyLiBLYXkgRGllZGVyaWNoczEtMCsGCSqGSIb3DQEJARYea2F5LmRpZWRlcmljaHNA >> dW5pLWtvbnN0YW56LmRlMIIBIjANBgkqhkiG9w0BAQEFAAOCAQ8AMIIBCgKCAQEAq8l >> +Wnxi >> cqMo9KicOd1kqdaq3pAymwWO/ >> J7ejbVjKChe10XnaITas8R1k9FrF54FsvssThDwmwsDWFjh >> eE0Fyegt+fk2tpXX4YnjqUcrlHkO5YHGQ0ogcrhvaMEKYoKLriH/ >> Nip0o8ljtMesL33gSPeV >> BOpyFwngqYUCt0rZPBytI+wxpN+SAEAO6erxvC4pASYpGTU5KBYPi5o5yf >> +9damc3flRF6Jl >> tbX+5aBFkra8CiWjH3q44DnnPn/DI+0HZs5goQWPmvzLA5GMy5T >> +3oXCGOKNFYFp7Y2AFdDf >> 2NyqSAo5KSd1D5qhCGWVedTeot435M5lqYWPJDFJj8Ew5QIDAQABo4IB/ >> jCCAfowgZcGCCsG >> AQUFBwEBBIGKMIGHMFEGCCsGAQUFBzAChkVodHRwOi8vd3d3LnRydXN0Y2VudGVyLmRlL2Nl >> cnRzZXJ2aWNlcy9jYWNlcnRzL3RjX2NsYXNzMV9MMV9DQV9WSS5jcnQwMgYIKwYBBQUHMAGG >> Jmh0dHA6Ly9vY3NwLlZJLnRjY2xhc3MxLnRydXN0Y2VudGVyLmRlMB8GA1UdIwQYMBaAFE6b >> 9bL73Wpr3EoHr8m7ZP8WSyafMAwGA1UdEwEB/wQCMAAwSgYDVR0gBEMwQTA/ >> BgkqghQALAEB >> AQEwMjAwBggrBgEFBQcCARYkaHR0cDovL3d3dy50cnVzdGNlbnRlci5kZS9ndWlkZWxpbmVz >> MA4GA1UdDwEB/wQEAwIE8DAdBgNVHQ4EFgQUac6i >> +TKOEt89dgE1SZwkDGCIPEcwVAYDVR0f >> BE0wSzBJoEegRYZDaHR0cDovL2NybC5WSS50Y2NsYXNzMS50cnVzdGNlbnRlci5kZS9jcmwv >> djIvdGNfY2xhc3MxX0wxX0NBX1ZJLmNybDAzBgNVHSUELDAqBggrBgEFBQcDAgYIKwYBBQUH >> AwQGCCsGAQUFBwMHBgorBgEEAYI3FAICMCkGA1UdEQQiMCCBHmtheS5kaWVkZXJpY2hzQHVu >> aS1rb25zdGFuei5kZTANBgkqhkiG9w0BAQUFAAOBgQBxO9sd6s4Lh9a2S70xvqvPkMZE6im >> + >> kI61FLhAhJqOqY3okJuO6xIdMjgbwblAuoEkHRaDTSna/2clqwH1k/ >> PiHn0TLHwWRCLIW3iO >> WjwkIewI1Jn3xsmxrvc3TLec69BcKHDwcsuE2rSv2GzM9qH0Vx7EuVehRrCa1oZNgQsEZzCC >> BN0wggRGoAMCAQICDwDGKQABAAIuCQlu6Nl5zDANBgkqhkiG9w0BAQUFADB8MQswCQYDVQQG >> EwJERTEcMBoGA1UEChMTVEMgVHJ1c3RDZW50ZXIgR21iSDElMCMGA1UECxMcVEMgVHJ1c3RD >> ZW50ZXIgQ2xhc3MgMSBMMSBDQTEoMCYGA1UEAxMfVEMgVHJ1c3RDZW50ZXIgQ2xhc3MgMSBM >> MSBDQSBWSTAeFw0wOTA2MTEwNzMzNTVaFw0xMDA2MTIwNzMzNTVaMFkxCzAJBgNVBAYTAkRF >> MRswGQYDVQQDExJEci4gS2F5IERpZWRlcmljaHMxLTArBgkqhkiG9w0BCQEWHmtheS5kaWVk >> ZXJpY2hzQHVuaS1rb25zdGFuei5kZTCCASIwDQYJKoZIhvcNAQEBBQADggEPADCCAQoCggEB >> AKvJflp8YnKjKPSonDndZKnWqt6QMpsFjvye3o21YygoXtdF52iE2rPEdZPRaxeeBbL7LE4Q >> 8JsLA1hY4XhNBcnoLfn5NraV1+GJ46lHK5R5DuWBxkNKIHK4b2jBCmKCi64h/ >> zYqdKPJY7TH >> rC994Ej3lQTqchcJ4KmFArdK2TwcrSPsMaTfkgBADunq8bwuKQEmKRk1OSgWD4uaOcn/ >> vXWp >> nN35UReiZbW1/uWgRZK2vAolox96uOA55z5/wyPtB2bOYKEFj5r8ywORjMuU/ >> t6FwhjijRWB >> ae2NgBXQ39jcqkgKOSkndQ+aoQhllXnU3qLeN+TOZamFjyQxSY/ >> BMOUCAwEAAaOCAf4wggH6 >> MIGXBggrBgEFBQcBAQSBijCBhzBRBggrBgEFBQcwAoZFaHR0cDovL3d3dy50cnVzdGNlbnRl >> ci5kZS9jZXJ0c2VydmljZXMvY2FjZXJ0cy90Y19jbGFzczFfTDFfQ0FfVkkuY3J0MDIGCCsG >> AQUFBzABhiZodHRwOi8vb2NzcC5WSS50Y2NsYXNzMS50cnVzdGNlbnRlci5kZTAfBgNVHSME >> GDAWgBROm/Wy+91qa9xKB6/Ju2T/ >> FksmnzAMBgNVHRMBAf8EAjAAMEoGA1UdIARDMEEwPwYJ >> KoIUACwBAQEBMDIwMAYIKwYBBQUHAgEWJGh0dHA6Ly93d3cudHJ1c3RjZW50ZXIuZGUvZ3Vp >> ZGVsaW5lczAOBgNVHQ8BAf8EBAMCBPAwHQYDVR0OBBYEFGnOovkyjhLfPXYBNUmcJAxgiDxH >> MFQGA1UdHwRNMEswSaBHoEWGQ2h0dHA6Ly9jcmwuVkkudGNjbGFzczEudHJ1c3RjZW50ZXIu >> ZGUvY3JsL3YyL3RjX2NsYXNzMV9MMV9DQV9WSS5jcmwwMwYDVR0lBCwwKgYIKwYBBQUHAwIG >> CCsGAQUFBwMEBggrBgEFBQcDBwYKKwYBBAGCNxQCAjApBgNVHREEIjAggR5rYXkuZGllZGVy >> aWNoc0B1bmkta29uc3RhbnouZGUwDQYJKoZIhvcNAQEFBQADgYEAcTvbHerOC4fWtku9Mb6r >> z5DGROopvpCOtRS4QISajqmN6JCbjusSHTI4G8G5QLqBJB0Wg00p2v9nJasB9ZPz4h59Eyx8 >> FkQiyFt4jlo8JCHsCNSZ98bJsa73N0y3nOvQXChw8HLLhNq0r9hszPah9FcexLlXoUawmtaG >> TYELBGcxggO0MIIDsAIBATCBjzB8MQswCQYDVQQGEwJERTEcMBoGA1UEChMTVEMgVHJ1c3RD >> ZW50ZXIgR21iSDElMCMGA1UECxMcVEMgVHJ1c3RDZW50ZXIgQ2xhc3MgMSBMMSBDQTEoMCYG >> A1UEAxMfVEMgVHJ1c3RDZW50ZXIgQ2xhc3MgMSBMMSBDQSBWSQIPAMYpAAEAAi4JCW7o2XnM >> MAkGBSsOAwIaBQCgggH5MBgGCSqGSIb3DQEJAzELBgkqhkiG9w0BBwEwHAYJKoZIhvcNAQkF >> MQ8XDTA5MDczMDE4NTYxNFowIwYJKoZIhvcNAQkEMRYEFFHzcLuH >> +jTkW7daRwRBDYwU1P54 >> MFIGCSqGSIb3DQEJDzFFMEMwCgYIKoZIhvcNAwcwDgYIKoZIhvcNAwICAgCAMA0GCCqGSIb3 >> DQMCAgFAMAcGBSsOAwIHMA0GCCqGSIb3DQMCAgEoMIGgBgkrBgEEAYI3EAQxgZIwgY8wfDEL >> MAkGA1UEBhMCREUxHDAaBgNVBAoTE1RDIFRydXN0Q2VudGVyIEdtYkgxJTAjBgNVBAsTHFRD >> IFRydXN0Q2VudGVyIENsYXNzIDEgTDEgQ0ExKDAmBgNVBAMTH1RDIFRydXN0Q2VudGVyIENs >> YXNzIDEgTDEgQ0EgVkkCDwDGKQABAAIuCQlu6Nl5zDCBogYLKoZIhvcNAQkQAgsxgZKggY8w >> fDELMAkGA1UEBhMCREUxHDAaBgNVBAoTE1RDIFRydXN0Q2VudGVyIEdtYkgxJTAjBgNVBAsT >> HFRDIFRydXN0Q2VudGVyIENsYXNzIDEgTDEgQ0ExKDAmBgNVBAMTH1RDIFRydXN0Q2VudGVy >> IENsYXNzIDEgTDEgQ0EgVkkCDwDGKQABAAIuCQlu6Nl5zDANBgkqhkiG9w0BAQEFAASCAQAw >> 8wlcwpSWk0jXsjHcylqYlQvIrFcJ02zUaEjW+r0aHNmRmS62v/ >> ILa8BVMaWc7j4rInYjcRQ/ >> 4MeBwqMOD7iK2usWIvp32SDnYdNY5qE4Jhn6F0eOLEpOZgET59XMYtzgBeMz2x/ >> fZUxDRxEo >> 0HeTHXjSv+kHeaPOVgkhoT5MbsV3Qvr7aBPW+yYO97Ek4FKxWX0AtgmqcWiK >> +hGTaxX5vELw >> ux4fjVWHKz66uovDStrp1Pgla0SIjbWyq7GYV0ol4Xq13r2IQgsR7SNV5hqFXSKT >> +M0YI44y >> I2Hfyc46XJ0pW9KFOO4DNxyLykju8ZdTC9OHSu503jsGiaJQTK/6AAAAAAAA >> --------------ms000208020902090005010407-- >> >> ------------------------------ >> >> Date: Thu, 30 Jul 2009 21:13:45 +0200 >> From: Luca Jovine <[email protected]> >> Subject: Postdoctoral position in protein crystallography at >> Karolinska Institutet >> >> POSTDOCTORAL FELLOWSHIP IN PROTEIN CRYSTALLOGRAPHY AT KAROLINSKA >> INSTITUT= >> ET >> >> As part of a collaboration between the laboratories of Rune >> Toftg=E5rd an= >> d Luca Jovine at the = >> >> KI Department of Biosciences and Nutrition and the Center for >> Biosciences= >> =2C a postdoctoral = >> >> position is immediately available on structural studies of key >> players in= >> the Hedgehog = >> >> signaling pathway=2E >> >> The Department of Biosciences and Nutrition has state-of-the-art >> equipmen= >> t for protein = >> >> expression=2C purification and in house X-ray data collection=3B >> frequent= >> access to synchrotron = >> >> sites and excellent computational facilities are also available=2E >> With t= >> heir close integration of = >> >> academic=2C clinical and industrial research=2C KI and the Center >> for Bio= >> sciences offer a highly = >> >> international and collaborative environment for biomedical studies=2E >> >> Highly motivated candidates with experience in protein expression >> (partic= >> ularly in insect = >> >> and/or mammalian cells)=2C purification=2C crystallization and >> structure = >> determination=2C are = >> >> encouraged to apply by e-mailing a curriculum vitae=2C summary of >> researc= >> h experience and = >> >> interests=2C and names and contact information for two references to >> Luca= >> Jovine = >> >> (luca=2Ejovine=40ki=2Ese)=2E Please note that=2C in order to be >> eligible = >> for this scholarship=2C candidates = >> >> should be non-Swedish citizens who have obtained a doctorate or the >> equiv= >> alent outside = >> >> Sweden=2E Deadline for application is 1 October 2009=2E >> >> ------------------------------------------------ >> Luca Jovine=2C Ph=2ED=2E >> Group Leader=2C Protein Crystallography Unit >> Karolinska Institutet >> Department of Biosciences and Nutrition >> H=E4lsov=E4gen 7=2C SE-141 57 Huddinge=2C Sweden >> Voice=3A +46=2E(0)8=2E6083-301 FAX=3A +46=2E(0)8=2E6089-290 >> E-mail=3A luca=2Ejovine=40ki=2Ese >> W3=3A http=3A//jovinelab=2Eorg >> ------------------------------------------------ >> >> ------------------------------ >> >> Date: Thu, 30 Jul 2009 12:29:09 -0700 >> From: Donnie Berkholz <[email protected]> >> Subject: Re: question of extra high B factor >> >> --/9DWx/yDrRhgMJTb >> Content-Type: text/plain; charset=us-ascii >> Content-Disposition: inline >> Content-Transfer-Encoding: quoted-printable >> >> On 00:00 Fri 31 Jul , Jiamu Du wrote: >>> Dear All, >>> I am refining a structure of a complex between of 50kD protein and >>> a 20kD >>> glycosylated protien. The data is of 2.9 A resolution. The wilson B >>> factor >>> is as high as 86.3 A^2. >>> The refinement seems well with R/Rf of 0.21/0.25. But the B-factor >>> is ext= >> ra >>> high. For the 50kD part, the average B factor is 76.5 A^2. But the >>> B fact= >> or >>> of the 20 kD glycosylated protein is as high as 133.3 A^2. Although >>> the >>> electron density looks fine, even the sugar chain is seen clearly. >>> My question is: >>> 1. How to reduce the B factor to a reasonable level? >>> 2. If it can not be redueced, when I published it, is this value >>> acceptab= >> le? >>> 3. In the same of similar resolutionIs, is there some other >>> structures li= >> ke >>> this situation? A component or a subunit of the protein has a >>> extra high= >> B >>> factor as high as 130. >> >> Our group published a paper a few years ago with an average B of 80 >> A^2=20 >> (JMB 328:893 (2003)). One referee had some objections to this, so >> we=20 >> performed analyses of the whole Protein Data Bank for proteins at=20 >> 2.5-3.0 A (see Fig. 2D in the paper). You may find this figure >> useful if=20 >> you need to convince anyone. >> >> --=20 >> Thanks, >> Donnie >> >> Donnie Berkholz >> P. Andrew Karplus lab >> Oregon State University >> >> --/9DWx/yDrRhgMJTb >> Content-Type: application/pgp-signature >> Content-Disposition: inline >> >> -----BEGIN PGP SIGNATURE----- >> Version: GnuPG v2.0.11 (GNU/Linux) >> >> iEYEABECAAYFAkpx9IUACgkQXVaO67S1rtvQfgCbBMtImQXESgPibA1Jy3ud4YP8 >> oCwAn1UG7SWOlclYAVw3eNRL3LHTqTe/ >> =gKGd >> -----END PGP SIGNATURE----- >> >> --/9DWx/yDrRhgMJTb-- >> >> ------------------------------ >> >> Date: Thu, 30 Jul 2009 17:53:15 -0400 >> From: Jim Fairman <[email protected]> >> Subject: Re: OpenGL Stereo 3D on 120 Hz LCDs, at last! >> >> --0015174be078b3c278046ff354f1 >> Content-Type: text/plain; charset=ISO-8859-1 >> Content-Transfer-Encoding: 7bit >> >> Just got this working on a machine with a Quadro FX 3800. Stereo >> looks >> great on both Pymol (v1.1) and the latest build of WinCoot (v0.6 build >> 2172). >> >> On Fri, Jul 17, 2009 at 1:20 AM, Warren DeLano <[email protected]> >> wrote: >> >>> FYI, for folks not subscribed to pymol-users: >>> >>> >>> >>> nVidia today released beta drivers which at last enable OpenGL- >>> based stereo >>> 3D visualization on 120 Hz LCDs using Quadro graphics cards. So >>> long as you >>> are willing to put up with Windows, you can finally abandon those >>> old CRTs >>> without spending a fortune and without sacrificing quality of the >>> stereo 3D >>> effect. >>> >>> >>> >>> Details posted at http://www.pymol.org >>> >>> >>> >>> Cheers, >>> >>> Warren >>> >>> >>> >> >> >> >> -- >> Jim Fairman >> Graduate Research Assistant >> Department of Biochemistry, Cellular, and Molecular Biology (BCMB) >> University of Tennessee -- Knoxville >> 216-368-3337 [email protected] [email protected] >> >> --0015174be078b3c278046ff354f1 >> Content-Type: text/html; charset=ISO-8859-1 >> Content-Transfer-Encoding: quoted-printable >> >> Just got this working on a machine with a Quadro FX 3800.=A0 Stereo >> looks g= >> reat on both Pymol (v1.1) and the latest build of WinCoot (v0.6 >> build 2172)= >> .<br><br><div class=3D"gmail_quote">On Fri, Jul 17, 2009 at 1:20 AM, >> Warren= >> DeLano <span dir=3D"ltr"><<a >> href=3D"mailto:[email protected]";>war...@d= >> elsci.com</a>></span> wrote:<br> >> <blockquote class=3D"gmail_quote" style=3D"border-left: 1px solid >> rgb(204, = >> 204, 204); margin: 0pt 0pt 0pt 0.8ex; padding-left: 1ex;"> >> >> >> >> >> >> >> >> >> >> >> >> <div link=3D"blue" vlink=3D"purple" lang=3D"EN-US"> >> >> <div> >> >> <p><font size=3D"2" face=3D"Arial"><span style=3D"font-size: 10pt; >> font-fam= >> ily: Arial;">FYI, for folks not subscribed to pymol-users:=A0 </ >> span></font= >>> </p> >> >> <p><font size=3D"2" face=3D"Arial"><span style=3D"font-size: 10pt; >> font-fam= >> ily: Arial;">=A0</span></font></p> >> >> <p><font size=3D"2" face=3D"Arial"><span style=3D"font-size: 10pt; >> font-fam= >> ily: Arial;">nVidia today released beta drivers which at last enable >> OpenGL= >> -based >> stereo 3D visualization on 120 Hz LCDs using Quadro graphics >> cards.=A0 So l= >> ong >> as you are willing to put up with Windows, you can finally abandon >> those ol= >> d CRTs >> without spending a fortune and without sacrificing quality of the >> stereo 3D >> effect.</span></font></p> >> >> <p><font size=3D"2" face=3D"Arial"><span style=3D"font-size: 10pt; >> font-fam= >> ily: Arial;">=A0</span></font></p> >> >> <p><font size=3D"2" face=3D"Arial"><span style=3D"font-size: 10pt; >> font-fam= >> ily: Arial;">Details posted at <a href=3D"http://www.pymol.org/"; >> target=3D"= >> _blank">http://www.pymol.org</a></span></font></p> >> >> <p><font size=3D"2" face=3D"Arial"><span style=3D"font-size: 10pt; >> font-fam= >> ily: Arial;">=A0</span></font></p> >> >> <p><font size=3D"2" face=3D"Arial"><span style=3D"font-size: 10pt; >> font-fam= >> ily: Arial;">Cheers,</span></font></p> >> >> <p><font size=3D"2" face=3D"Arial"><span style=3D"font-size: 10pt; >> font-fam= >> ily: Arial;">Warren</span></font><font size=3D"2" >> face=3D"Arial"><span styl= >> e=3D"font-size: 10pt; font-family: Arial;"></span></font></p> >> >> <p><font size=3D"2" face=3D"Arial"><span style=3D"font-size: 10pt; >> font-fam= >> ily: Arial;">=A0</span></font></p> >> >> </div> >> >> </div> >> >> >> </blockquote></div><br><br clear=3D"all"><br>-- <br>Jim >> Fairman<br>Graduate= >> Research Assistant<br>Department of Biochemistry, Cellular, and >> Molecular = >> Biology (BCMB)<br>University of Tennessee -- >> Knoxville<br>216-368-3337 <a h= >> ref=3D"mailto:[email protected]";>[email protected]</a> <a >> href=3D"mailto:jame= >> [email protected]">[email protected]</a><br> >> >> >> --0015174be078b3c278046ff354f1-- >> >> ------------------------------ >> >> Date: Thu, 30 Jul 2009 15:23:04 -0700 >> From: James Holton <[email protected]> >> Subject: Re: Foils for energy calibration >> >> At ALS, we have a box of foils from EXAFS Materials that seems to >> get=20 >> passed around from beamline to beamline. I ran absorption scans on >> 17=20 >> edges from the metals in the box one day, and found that there was=20 >> considerable scatter in the expected vs observed edge positions: >> http://bl831.als.lbl.gov/~jamesh/pickup/mono_calib.png >> Here I have plotted the "correct" position of each edge as >> determined by=20 >> Bearden & Burr (1967), against the edge I determined using the >> criterion=20 >> recommended in the "Reference Spectra" document in the=20 >> exafsmaterials.com website: the first inflection point in the >> derivative=20 >> spectrum. I think a large amount of the scatter is because my mono=20 >> (like many PX/MX beamlines) is Si(111) and not Si(220) like the one >> used=20 >> to determine the reference spectra. It is not hard to imagine how=20 >> blurring the spectrum with a wider energy spread of the incident >> beam=20 >> will shift the position of the "edge". One could try to use the=20 >> electron binding energy tabulated in the "little orange book": >> http://xdb.lbl.gov/Section1/Sec_1-1.html >> but these do not always take into account the "near edge" features >> (like=20 >> the white line from SeMet) which change depending on the chemical=20 >> environment around the metal, radiation damage, etc. It would be >> nice=20 >> if someone could calibrate some standard reference materials using >> a=20 >> Si(111) monochromator, but I don't know of anyone who has done this. >> >> >> However, another way to get your x-ray wavelength is using Bragg's >> law: >> lambda =3D 2*d*sin(theta) >> and the d-spacing of silicon is known to be 5.43159 =B1 0.00020 A, >> and=20 >> NIST will sell you certified Si powder: >> https://www-s.nist.gov/srmors/view_detail.cfm?srm=3D640d >> >> The problem here is that although you know d very accurately, the >> error=20 >> in lambda is dominated by sin(theta), or rather the uncertainty in >> your=20 >> detector distance. The pixel field on most detectors is actually >> quite=20 >> accurate, as a NIST-traceable calibration is used to make the >> pinhole=20 >> calibration mask, and the encoder on most detector distance stages >> is=20 >> very accurate for relative moves (counting ticks on the encoder). >> But=20 >> there is always an offset from the "zero" position predicted by the=20 >> encoder to the true center of rotation that is hard to know.=20 >> >> Nevertheless, all you really want is for the d-spacing of silicon >> powder=20 >> rings to be right at all detector distances. You can use the >> program=20 >> FIT2D to refine the wavelength, distance, detector tilt, etc. or >> any=20 >> combination thereof for a given image, but you will find that the=20 >> repeatability of such a fit (using different starting parameters) is >> not=20 >> great because the distance and wavelength are highly correlated. =20 >> However, there is a way around this: >> >> Since we know that a relative move of the distance will be >> accurate,=20 >> there should be one and only one offset that you can add to the >> recorded=20 >> value of distance of each image to make it the "right" distance. >> You=20 >> can define the "right" offset as the one where FIXing the resulting=20 >> "right" distance in FIT2D and refining everything else gives you >> the=20 >> same refined value for the wavelength from every image. You need >> to=20 >> manually "refine" this offset for a few rounds. What you will >> generally=20 >> see is that the graph of fitted wavelength vs the distance is a >> straight=20 >> line, and you want to make the slope of this line to be zero. =20 >> Eventually, you will arrive at some offset that gives you the >> smallest=20 >> spread in refined wavelength values. The average refined wavelength >> is=20 >> then the "true" wavelength. Should be able to get it within one or >> two=20 >> eV. Perhaps more if you take a lot of silicon powder images. >> >> At ALS beamlines 8.3.1 and 12.3.1 I have done both kinds of >> calibration,=20 >> and I am fairly certain I get the wavelength accurate to within 1 eV >> by=20 >> calibrating the half-way-up point of an absorption scan of a ~122 >> micron=20 >> thick copper metal foil to 8979.0 eV. >> >> -James Holton >> MAD Scientist >> >> >> Richard Gillilan wrote: >>> In the past we've used elemental foils from exafsmaterials.com for=20 >>> energy calibration of our MAD beamline. These standards are for >>> EXAFS=20 >>> and XANES. Most are thin (5 micron) metal foils. >>> >>> Has anyone had experience with other sources of standards or other=20 >>> forms (such as compounds rather than pure elements)? >>> >>> I notice that a number of companies offer XRF standard kits. >>> >>> Richard Gillilan >>> MacCHESS >> >> ------------------------------ >> >> End of CCP4BB Digest - 29 Jul 2009 to 30 Jul 2009 (#2009-207) >> ************************************************************* >
{kind=link}