Hi, Thank you all for the answers.
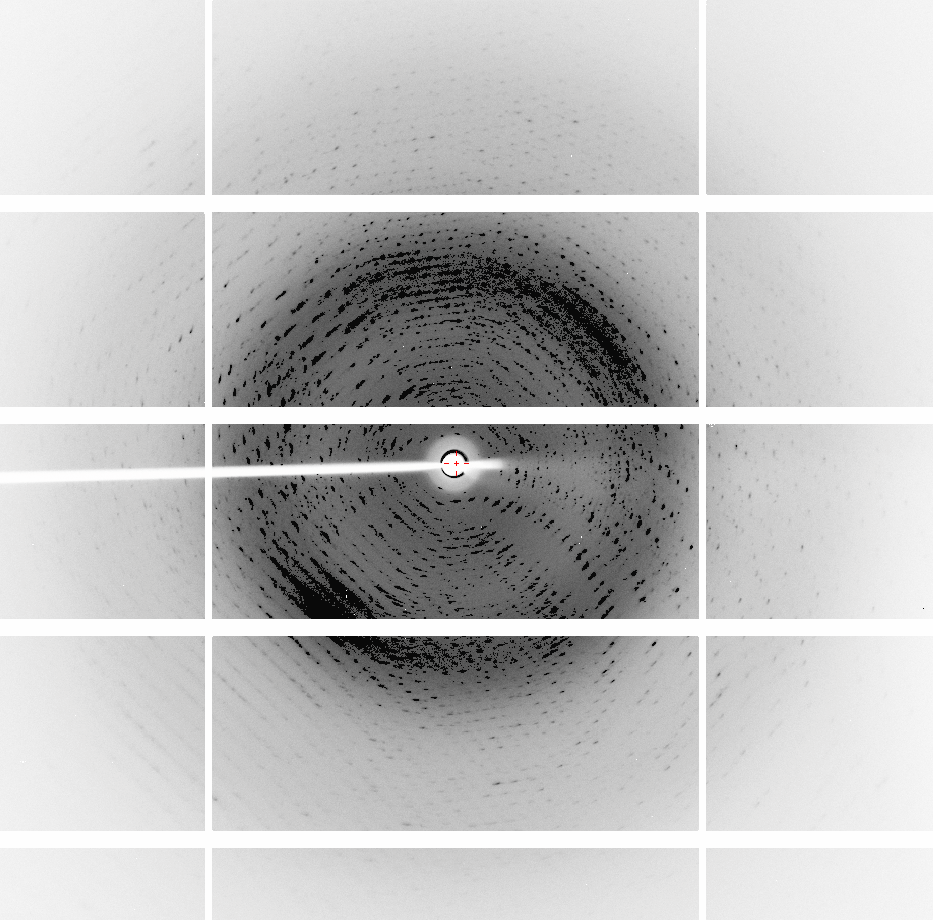
Eleanor, you nailed it, thank you! Your observation that the cell in P6 had a different C axis led me to reprocess the data using a cell of the same size of the other space groups. I have commented that processing in P6 before yielded r_free > 0.50, but now the r_free is 0.3232 without using a twin law, and using the twin law h,-h-k,-l which was recommended by Phenix Xtriage, yields r_work = 0.1674 and r_free = 0.1960. Thank you for the other suggestions and reference, I followed them. I reviewed all my analyses and only P6 had the wrong cell size. I don't know why. - P3 yields r_work = 0.1711 and r_free = 0.1935 with 120 water molecules (the asymmetric unit contains 420 residues and the resolution is 2.01 A). - P6 yields r_work = 0.1674 and r_free = 0.1960 with 94 water molecules (the asymmetric unit contains 210 residues and the resolution is 1.91 A). I guess I should stick with P6, since it has higher symmetry, right? Regardless of how I process the data, in all space groups, pointless always says it is sure that the space group P622. That is probably due to the twinning, that causes reflections to lay over each other and gives the appearance of higher symmetry. A question, does it makes sense to use TLS and a twin law to refine the data at this resolution (1.9 A)? Carlos, thank you for the references. Carlos Frazao, the PDB header generated by Phenix indicates that the twin fraction is still 0.490 after refinement in P3 and P6. Jacob Keller, I have tried all trigonal and hexagonal space groups, except for those I reported (P1, P3 and now P6), all others yield unrefinable solutions (R_free > 0.41). However, there are a lot of explanations for it, for example, the data might be processed using the wrong cell size, or the MR solution might just be wrong. Thank you all for the help! I'm still looking for references that can teach me how to look at protein diffraction images and understand what I am seeing. The basics like recognizing bad data and what usually leads to deformity in the spots (for example, elliptical or duplicated) would be of great help. Regards, Napo ----- Mensagem original ----- > De: "Eleanor Dodson" <[email protected]> > Para: [email protected] > Enviadas: Quarta-feira, 30 de Novembro de 2016 14:10:16 > Assunto: Re: [ccp4bb] To win or not twinning? > First - there is a useful document about possible twin laws in the > CCP4 documentation. It references the SHELX description of twinning > for small molecules. > http://www.ccp4.ac.uk/html/twinning.html > It is possible in a trigonal system that you could have two twin > operators but the twinning stats should lok odd in that case > For a trigonal space group there are several options, but your > technique seems to have correctly found SG P3. > As Jacob Keller says - check POINTLESS to see if P322 etc are > options? > Or ask Zanudu.. > Just to comment - I think you must have the cell dimensions wrong for > the P63 case? I cant see how the c axis can be longer? > I like the data processing POINTLESS/AIMLESS/ CTRUNCATE.. > POINTLESS lists all axial reflections so you can make an educated > guess at screw axes (or let POINTLESS do it for you...) > POINTLESS analyses each symmetry operator separately. So if the -k, > -h, -l was slightly less convincing than others, AND the twin tests > suggested twinning, you might expect that to be a twin operator and > not a symmetry operator.. > ctruncate tests each possible twin operator and scores them. (And so > does XTRIAGE of course..) > On 30 November 2016 at 14:00, Keller, Jacob < > [email protected] > wrote: > > What about spacegroups in PG 32, e.g., p3212? > > > JPK > > > From: CCP4 bulletin board [mailto: [email protected] ] On > > Behalf > > Of Napoleao Fonseca Valadares > > > Sent: Wednesday, November 30, 2016 2:01 AM > > > To: [email protected] > > > Subject: [ccp4bb] To win or not twinning? > > > Dear CCP4ers, > > > I'd like to kindly ask your advice. Sorry for the long e-mail. > > > I have crystals of a 12.3 KDa protein that grow in hexagon-like > > patterns, link for the crystal image: > > > http://fullonline.org/science/cryst01.jpg > > > XDS, Phenix and Pointless always suggest that the data sets for > > these > > crystals belong to the space group P622. However, Phenix, Phaser > > and > > Pointless indicate that twinning is present. > > > "Bad looking" diffraction images, diffracted to 1.6 A (collected 6 > > months ago): > > > http://fullonline.org/science/dataset1_image37.png > > > http://fullonline.org/science/dataset1_image7.png > > > Best data set, diffracted to 2.01 A (collected a month ago): > > > http://fullonline.org/science/dataset2_image51.png > > > The second data set present better looking images, a better XDS ISa > > value (around 24) and diffracted to 2.2 A. The "bad looking" data > > set diffracted to 1.6 A, but I decided to stop working with it (XDS > > ISa around 10). > > > There is a template with 60% identity, I used XDS to try to process > > the data in all trigonal/hexagonal space groups from P3 to P6(3)22, > > and spend a lot of time trying molecular replacement procedures in > > Phaser and Morda, and refining the candidate solutions. Used Zanuda > > too, trying to figure out the space group (and read as much as > > possible in this CCP4 list looking for similar cases). > > > Unit cells and typical MR results: > > > P1: 56.430 77.718 77.673 119.99 89.99 89.97 > > > SOLU SET RFZ=9.5 TFZ=* PAK=0 LLG=91 RF++ TFZ=18.2 PAK=0 LLG=311 > > TFZ==18.4 (& > > > TFZ==17.0) LLG+=(311 & 726) LLG=5751 TFZ==64.6 PAK=0 LLG=5751 > > TFZ==64.6 > > > P3: 77.675 77.675 56.409 90.00 90.00 120.00 > > > RFZ=5.5 TFZ=8.5 PAK=0 LLG=86 TFZ==9.4 LLG=2575 TFZ==46.5 > > > P6: 77.534 77.534 92.986 90.00 90.00 120.00 > > > RFZ=11.2 TFZ=16.2 PAK=2 LLG=437 TFZ==25.7 LLG=437 TFZ==25.7 > > > P6(3): 77.675 77.675 56.409 90.00 90.00 120.00 > > > RFZ=8.8 TFZ=11.5 PAK=0 LLG=66 TFZ==7.6 LLG=66 TFZ==7.6 > > > P622: 77.660 77.660 56.400 90.00 90.00 120.00 > > > RFZ=4.8 TFZ=5.4 PAK=1 LLG=22 TFZ==4.7 LLG=24 TFZ==4.8 > > > In P1 (LLG=5751 TFZ==64.6) there are 12 molecules in the asymmetric > > unit, and in P3 (LLG=2575 TFZ==46.5) 4 molecules. Packing looks > > good, in P1 the ASU looks like two superposed hexagonal donuts > > formed by 6 molecules each. > > > Refining in P1, without adding waters or TLS, yield r_work = 0.2964 > > and r_free = 0.3428, and it is hard to decrease these values. > > > Refining in P3, I managed to get r_work = 0.2934 and r_free = > > 0.3399, > > but looks like it's not getting any better than this. > > > Refining in P6 yields horrible r_free values (>0.50). > > > Trying to refine MR solutions in any other other space groups yield > > Rfree 0.41 or more. > > > If in the P3 space group I use the twin operator -k,-h,-l > > (estimated > > twin fraction of 0.490) suggested by Phenix Xtriage, the values > > miraculously go down to r_work = 0.1923 r_free = 0.2202, without > > waters (r_free without using a twin law = 0.3399). Adding 120 water > > molecules and doing some refining yields 0.1711 r_free = 0.1935 > > (the > > asymmetric unit contains 420 residues and the resolution is 2.01 > > A). > > > I've been reading about twinning refinement and how it can drop the > > Rvalues, but from what I understood if I use it improperly I may > > compromise the refinement quality. > > > I would like advice on: > > > 1 - References that can teach how to look at protein diffraction > > images and understand what I am seeing. The basics like recognizing > > bad data and what usually leads to deformity in the spots (for > > example, elliptical or duplicated) would be of great help. > > > 2 - Should I look for other space groups? What else could be tried? > > Is this a case where a twin law should be used in the refinement? > > If > > yes, what can I do to confirm the need for a twin law in the > > refinement? > > > Thank you all in advance. > > > Regards, > > > Napo >
{kind=link}
{kind=link}
{kind=link}
{kind=link}