Not sure about protein diffraction images - I would recommend going to any course where Zbysek Dauter was teaching - he is the master.. And he lectured at severa;l CCP4 Study Weekends. It would be worth looking up those. I think they are online at the CCP4 website or in Acta Cryst.
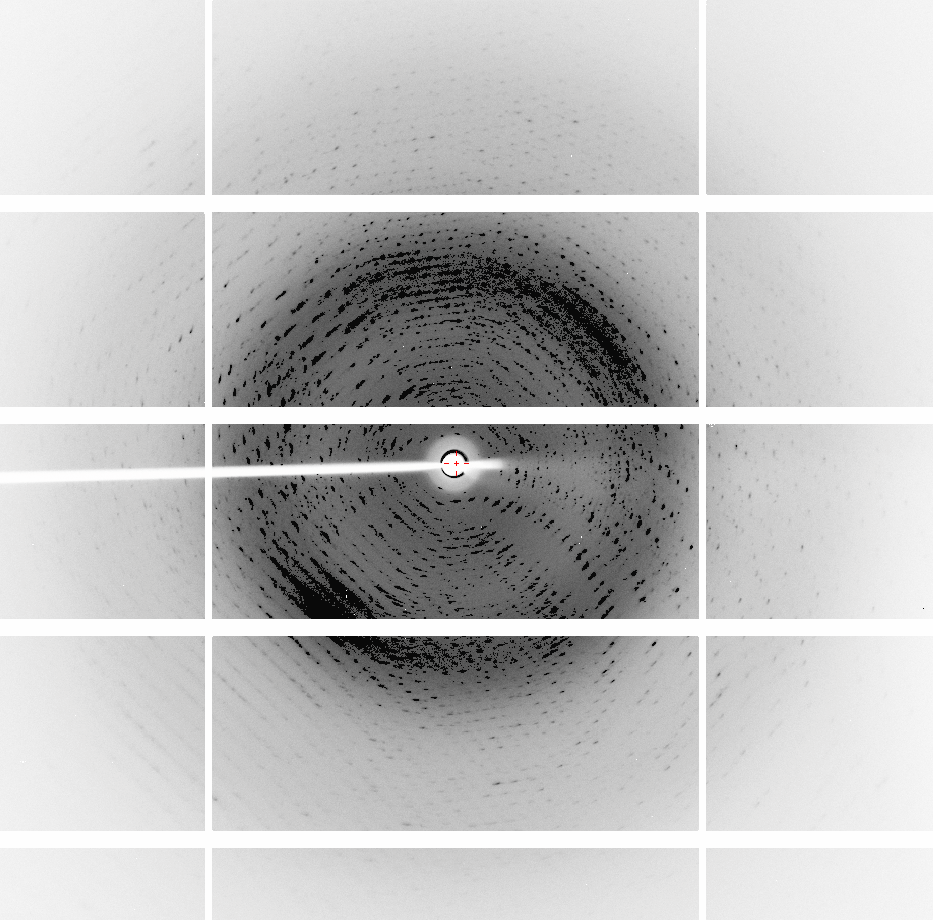
Eleanor On 1 December 2016 at 20:24, Napoleao Fonseca Valadares <[email protected]> wrote: > Hi, > > Thank you all for the answers. > > Eleanor, you nailed it, thank you! Your observation that the cell in P6 > had a different C axis led me to reprocess the data using a cell of the > same size of the other space groups. I have commented that processing in P6 > before yielded r_free > 0.50, but now the r_free is 0.3232 without using a > twin law, and using the twin law h,-h-k,-l which was recommended by Phenix > Xtriage, yields r_work = 0.1674 and r_free = 0.1960. Thank you for the > other suggestions and reference, I followed them. > > I reviewed all my analyses and only P6 had the wrong cell size. I don't > know why. > > - P3 yields r_work = 0.1711 and r_free = 0.1935 with 120 water molecules > (the asymmetric unit contains 420 residues and the resolution is 2.01 A). > - P6 yields r_work = 0.1674 and r_free = 0.1960 with 94 water molecules > (the asymmetric unit contains 210 residues and the resolution is 1.91 A). > > I guess I should stick with P6, since it has higher symmetry, right? > > Regardless of how I process the data, in all space groups, pointless > always says it is sure that the space group P622. That is probably due to > the twinning, that causes reflections to lay over each other and gives the > appearance of higher symmetry. > > A question, does it makes sense to use TLS and a twin law to refine the > data at this resolution (1.9 A)? > > Carlos, thank you for the references. > > Carlos Frazao, the PDB header generated by Phenix indicates that the twin > fraction is still 0.490 after refinement in P3 and P6. > > Jacob Keller, I have tried all trigonal and hexagonal space groups, except > for those I reported (P1, P3 and now P6), all others yield unrefinable > solutions (R_free > 0.41). However, there are a lot of explanations for it, > for example, the data might be processed using the wrong cell size, or the > MR solution might just be wrong. > > Thank you all for the help! > > I'm still looking for references that can teach me how to look at protein > diffraction images and understand what I am seeing. The basics like > recognizing bad data and what usually leads to deformity in the spots (for > example, elliptical or duplicated) would be of great help. > > Regards, > Napo > > > ------------------------------ > > *De: *"Eleanor Dodson" <[email protected]> > *Para: *[email protected] > *Enviadas: *Quarta-feira, 30 de Novembro de 2016 14:10:16 > *Assunto: *Re: [ccp4bb] To win or not twinning? > > > First - there is a useful document about possible twin laws in the CCP4 > documentation. It references the SHELX description of twinning for small > molecules. > http://www.ccp4.ac.uk/html/twinning.html > > It is possible in a trigonal system that you could have two twin operators > but the twinning stats should lok odd in that case > > For a trigonal space group there are several options, but your technique > seems to have correctly found SG P3. > As Jacob Keller says - check POINTLESS to see if P322 etc are options? > Or ask Zanudu.. > > Just to comment - I think you must have the cell dimensions wrong for the > P63 case? I cant see how the c axis can be longer? > > I like the data processing POINTLESS/AIMLESS/ CTRUNCATE.. > > POINTLESS lists all axial reflections so you can make an educated guess at > screw axes (or let POINTLESS do it for you...) > POINTLESS analyses each symmetry operator separately. So if the -k, -h, -l > was slightly less convincing than others, AND the twin tests suggested > twinning, you might expect that to be a twin operator and not a symmetry > operator.. > > ctruncate tests each possible twin operator and scores them. (And so does > XTRIAGE of course..) > > > On 30 November 2016 at 14:00, Keller, Jacob <[email protected]> > wrote: > >> What about spacegroups in PG 32, e.g., p3212? >> >> >> >> JPK >> >> >> >> *From:* CCP4 bulletin board [mailto:[email protected]] *On Behalf Of >> *Napoleao Fonseca Valadares >> *Sent:* Wednesday, November 30, 2016 2:01 AM >> *To:* [email protected] >> *Subject:* [ccp4bb] To win or not twinning? >> >> >> >> Dear CCP4ers, >> >> I'd like to kindly ask your advice. Sorry for the long e-mail. >> >> I have crystals of a 12.3 KDa protein that grow in hexagon-like patterns, >> link for the crystal image: >> http://fullonline.org/science/cryst01.jpg >> >> XDS, Phenix and Pointless always suggest that the data sets for these >> crystals belong to the space group P622. However, Phenix, Phaser and >> Pointless indicate that twinning is present. >> >> "Bad looking" diffraction images, diffracted to 1.6 A (collected 6 months >> ago): >> http://fullonline.org/science/dataset1_image37.png >> http://fullonline.org/science/dataset1_image7.png >> >> Best data set, diffracted to 2.01 A (collected a month ago): >> http://fullonline.org/science/dataset2_image51.png >> >> The second data set present better looking images, a better XDS ISa value >> (around 24) and diffracted to 2.2 A. The "bad looking" data set diffracted >> to 1.6 A, but I decided to stop working with it (XDS ISa around 10). >> >> There is a template with 60% identity, I used XDS to try to process the >> data in all trigonal/hexagonal space groups from P3 to P6(3)22, and spend a >> lot of time trying molecular replacement procedures in Phaser and Morda, >> and refining the candidate solutions. Used Zanuda too, trying to figure out >> the space group (and read as much as possible in this CCP4 list looking for >> similar cases). >> >> Unit cells and typical MR results: >> >> P1: 56.430 77.718 77.673 119.99 89.99 89.97 >> SOLU SET RFZ=9.5 TFZ=* PAK=0 LLG=91 RF++ TFZ=18.2 PAK=0 LLG=311 >> TFZ==18.4 (& >> TFZ==17.0) LLG+=(311 & 726) LLG=5751 TFZ==64.6 PAK=0 LLG=5751 >> TFZ==64.6 >> >> P3: 77.675 77.675 56.409 90.00 90.00 120.00 >> RFZ=5.5 TFZ=8.5 PAK=0 LLG=86 TFZ==9.4 LLG=2575 TFZ==46.5 >> >> P6: 77.534 77.534 92.986 90.00 90.00 120.00 >> RFZ=11.2 TFZ=16.2 PAK=2 LLG=437 TFZ==25.7 LLG=437 TFZ==25.7 >> >> P6(3): 77.675 77.675 56.409 90.00 90.00 120.00 >> RFZ=8.8 TFZ=11.5 PAK=0 LLG=66 TFZ==7.6 LLG=66 TFZ==7.6 >> >> P622: 77.660 77.660 56.400 90.00 90.00 120.00 >> RFZ=4.8 TFZ=5.4 PAK=1 LLG=22 TFZ==4.7 LLG=24 TFZ==4.8 >> >> >> In P1 (LLG=5751 TFZ==64.6) there are 12 molecules in the asymmetric unit, >> and in P3 (LLG=2575 TFZ==46.5) 4 molecules. Packing looks good, in P1 the >> ASU looks like two superposed hexagonal donuts formed by 6 molecules each. >> Refining in P1, without adding waters or TLS, yield r_work = 0.2964 and >> r_free = 0.3428, and it is hard to decrease these values. >> Refining in P3, I managed to get r_work = 0.2934 and r_free = 0.3399, but >> looks like it's not getting any better than this. >> Refining in P6 yields horrible r_free values (>0.50). >> >> Trying to refine MR solutions in any other other space groups yield Rfree >> 0.41 or more. >> >> If in the P3 space group I use the twin operator -k,-h,-l (estimated twin >> fraction of 0.490) suggested by Phenix Xtriage, the values miraculously go >> down to r_work = 0.1923 r_free = 0.2202, without waters (r_free without >> using a twin law = 0.3399). Adding 120 water molecules and doing some >> refining yields 0.1711 r_free = 0.1935 (the asymmetric unit contains 420 >> residues and the resolution is 2.01 A). >> I've been reading about twinning refinement and how it can drop the >> Rvalues, but from what I understood if I use it improperly I may compromise >> the refinement quality. >> >> I would like advice on: >> 1 - References that can teach how to look at protein diffraction images >> and understand what I am seeing. The basics like recognizing bad data and >> what usually leads to deformity in the spots (for example, elliptical or >> duplicated) would be of great help. >> >> 2 - Should I look for other space groups? What else could be tried? Is >> this a case where a twin law should be used in the refinement? If yes, what >> can I do to confirm the need for a twin law in the refinement? >> >> Thank you all in advance. >> Regards, >> Napo >> > > >
{kind=link}
{kind=link}
{kind=link}
{kind=link}