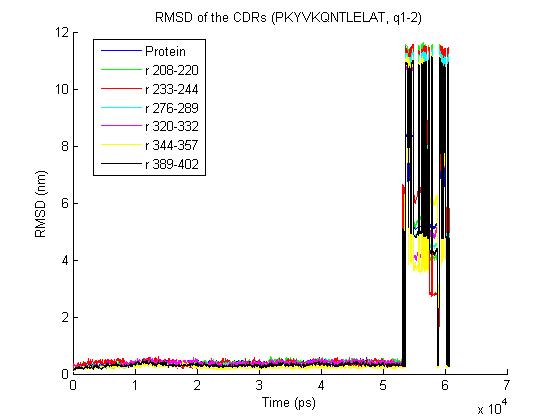
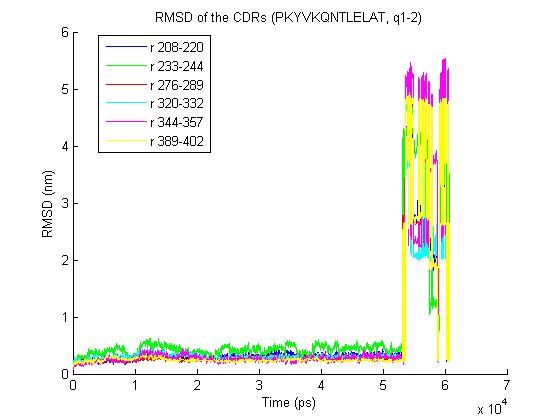
1) trjconv with the "-pbc nojump" option does not help - it removes most
of the jumps but one big remains- the rmsd for the trajectory is
http://www.meduniwien.ac.at/msi/biosim/bk/4.jpg
before the converting with -pbc and
http://www.meduniwien.ac.at/msi/biosim/bk/4noJump.jpg
after conversion.
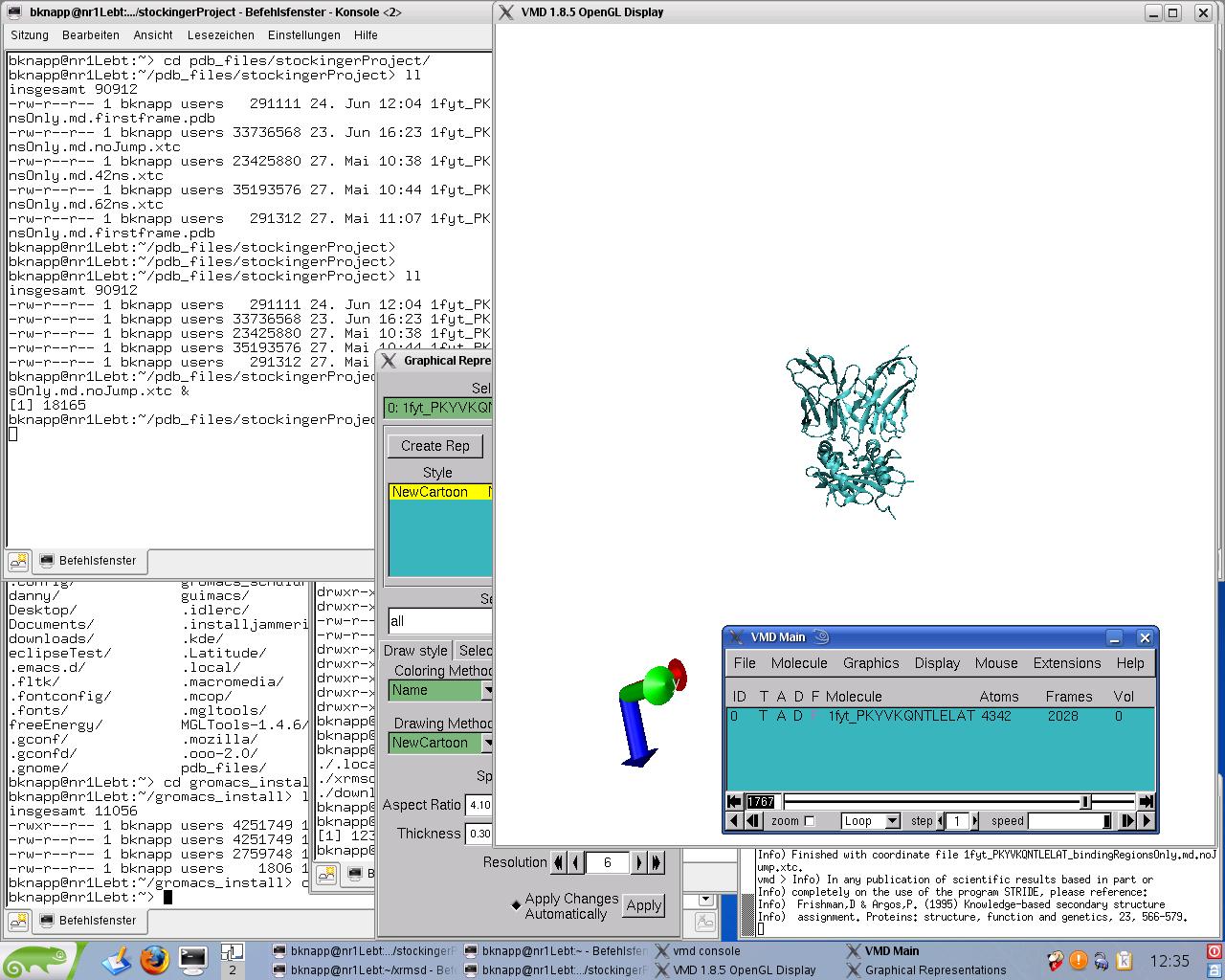
interestingly under linux vmd could load the trajectories which look
like that:
http://www.meduniwien.ac.at/msi/biosim/bk/4afterNoJump1767.jpg
and
http://www.meduniwien.ac.at/msi/biosim/bk/4afterNoJump1768.jpg
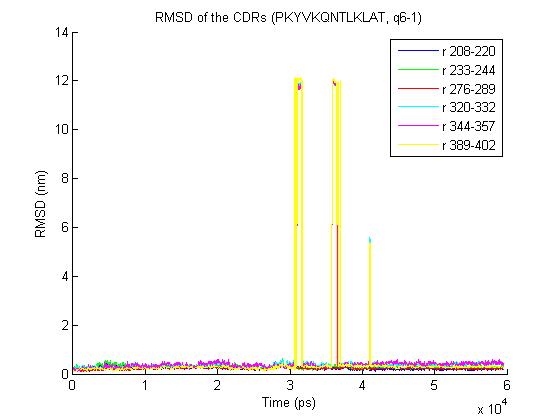
2) Here an illustration for other trajectories with the same problem:
http://www.meduniwien.ac.at/msi/biosim/bk/1.jpg
http://www.meduniwien.ac.at/msi/biosim/bk/2.jpg
http://www.meduniwien.ac.at/msi/biosim/bk/jumppoint.jpg
3) gmxcheck returns no error, just this:
trn version: GMX_trn_file (single precision)
Reading frame 0 time 0.000
# Atoms 167731
Reading frame 20000 time 60000.000
Item #frames Timestep (ps)
Step 20261 3
Time 20261 3
Lambda 20261 3
Coords 20261 3
Velocities 2027 30
Forces 2027 30
Box 20261 3
any further ideas what I could do with those trajectories or what I
shall do to avoid such problems in future (integration step, water shell
size, ... ?)
cheers
Bernhard
------------------------------
Message: 4
Date: Mon, 22 Jun 2009 16:37:44 +0200
From: Erik Marklund <[email protected]>
Subject: Re: [gmx-users] problems with some calculated trajectories
To: Discussion list for GROMACS users <[email protected]>
Message-ID: <[email protected]>
Content-Type: text/plain; charset=ISO-8859-1; format=flowed
Hi,
Could it be a periodicity issue? Have you tried using trjconv -pbc nojump ?
/Erik
------------------------------
Message: 2
Date: Mon, 22 Jun 2009 17:19:43 +0200
From: Martin H?fling <[email protected]>
Subject: Re: [gmx-users] problems with some calculated trajectories
To: Discussion list for GROMACS users <[email protected]>
Message-ID: <[email protected]>
Content-Type: text/plain; charset=US-ASCII; format=flowed; delsp=yes
Hey Bernhard, here some thoughts of mine:
In principle, vmd should be able to read the trajectories if pdb2gmx
tells you that the trajectory is ok. If this is not the case, you can
use gmx_rescue64 to extract the undamaged parts.
Reasons for damaged trajectories, even when gromacs runs through fine...
... for me - filesystem issues were causing this. In particular this
happened for me with NFS shares and on Lustre filesystems. My NFS
problems got solved with an upgrade of the NFS packages on our
machines (at least 1 1/2 yrs ago).
Best
Martin
------------------------------
Message: 3
Date: Mon, 22 Jun 2009 18:06:13 +0200
From: Martin H?fling <[email protected]>
Subject: Re: [gmx-users] problems with some calculated trajectories
To: Discussion list for GROMACS users <[email protected]>
Message-ID: <[email protected]>
Content-Type: text/plain; charset=ISO-8859-1; format=flowed; delsp=yes
Am 22.06.2009 um 17:19 schrieb Martin Höfling:
Hey Bernhard, here some thoughts of mine:
In principle, vmd should be able to read the trajectories if pdb2gmx
tells you that the trajectory is ok. If this is not the case, you
can use gmx_rescue64 to extract the undamaged parts.
^
^^^^^^^^^^
= gmxcheck :-)
Best
Martin
------------------------------
Message: 4
Date: Mon, 22 Jun 2009 13:29:10 -0400
From: "Justin A. Lemkul" <[email protected]>
Subject: Re: [gmx-users] -bash:
/home_soft/soft_chem/soft_install/gromac/bin/GMXRC.bash: No such
file
or directory
To: Discussion list for GROMACS users <[email protected]>
Message-ID: <[email protected]>
Content-Type: text/plain; charset=GB2312
_______________________________________________
gmx-users mailing list [email protected]
http://lists.gromacs.org/mailman/listinfo/gmx-users
Please search the archive at http://www.gromacs.org/search before posting!
Please don't post (un)subscribe requests to the list. Use the
www interface or send it to [email protected].
Can't post? Read http://www.gromacs.org/mailing_lists/users.php
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}