Hi Bernhard, The images, are they taken from exactly the same angle? If so, there's some change in orientation that is just impossible in such a short time. You didn't happen to fit your structure to a reference prior to removal of jumps, did you? Fitting messes up PBC and garbles results from -pbc nojump. If these are not from the original trajectory (mdrun output), have a look what happens there. Otherwise, trace back all your steps. Maybe paste here what you've done exactly...
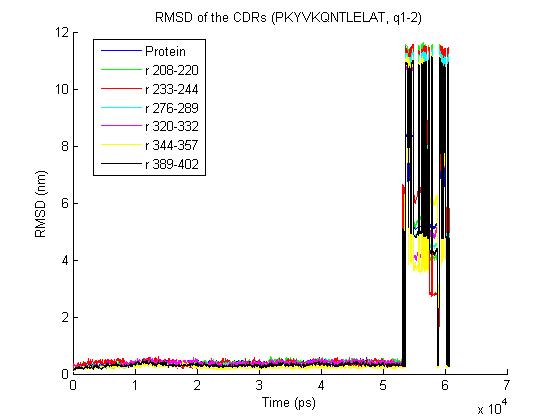
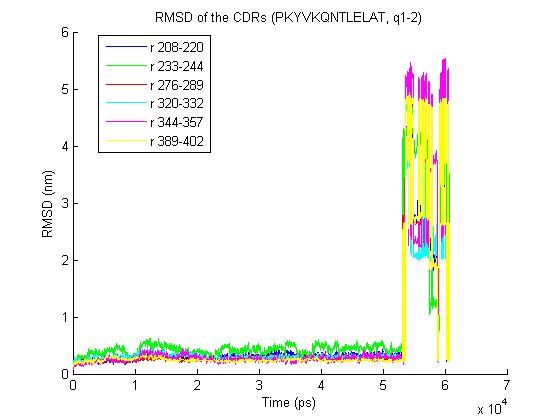
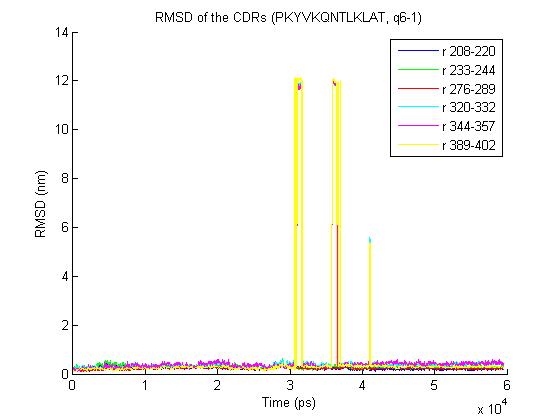
Cheers, Tsjerk On Thu, Jun 25, 2009 at 10:10 AM, Bernhard Knapp<[email protected]> wrote: > 1) trjconv with the "-pbc nojump" option does not help - it removes most of > the jumps but one big remains- the rmsd for the trajectory is > http://www.meduniwien.ac.at/msi/biosim/bk/4.jpg > before the converting with -pbc and > http://www.meduniwien.ac.at/msi/biosim/bk/4noJump.jpg > after conversion. > > interestingly under linux vmd could load the trajectories which look like > that: > http://www.meduniwien.ac.at/msi/biosim/bk/4afterNoJump1767.jpg > and > http://www.meduniwien.ac.at/msi/biosim/bk/4afterNoJump1768.jpg > > 2) Here an illustration for other trajectories with the same problem: > http://www.meduniwien.ac.at/msi/biosim/bk/1.jpg > http://www.meduniwien.ac.at/msi/biosim/bk/2.jpg > http://www.meduniwien.ac.at/msi/biosim/bk/jumppoint.jpg > > > 3) gmxcheck returns no error, just this: > > trn version: GMX_trn_file (single precision) > Reading frame 0 time 0.000 > # Atoms 167731 > Reading frame 20000 time 60000.000 > > Item #frames Timestep (ps) > Step 20261 3 > Time 20261 3 > Lambda 20261 3 > Coords 20261 3 > Velocities 2027 30 > Forces 2027 30 > Box 20261 3 > > > any further ideas what I could do with those trajectories or what I shall do > to avoid such problems in future (integration step, water shell size, ... ?) > > cheers > Bernhard > > > > > > > ------------------------------ > > Message: 4 > Date: Mon, 22 Jun 2009 16:37:44 +0200 > From: Erik Marklund <[email protected]> > Subject: Re: [gmx-users] problems with some calculated trajectories > To: Discussion list for GROMACS users <[email protected]> > Message-ID: <[email protected]> > Content-Type: text/plain; charset=ISO-8859-1; format=flowed > > Hi, > > Could it be a periodicity issue? Have you tried using trjconv -pbc nojump ? > > /Erik > > ------------------------------ > > Message: 2 > Date: Mon, 22 Jun 2009 17:19:43 +0200 > From: Martin H?fling <[email protected]> > Subject: Re: [gmx-users] problems with some calculated trajectories > To: Discussion list for GROMACS users <[email protected]> > Message-ID: <[email protected]> > Content-Type: text/plain; charset=US-ASCII; format=flowed; delsp=yes > > Hey Bernhard, here some thoughts of mine: > > In principle, vmd should be able to read the trajectories if pdb2gmx > tells you that the trajectory is ok. If this is not the case, you can > use gmx_rescue64 to extract the undamaged parts. > > Reasons for damaged trajectories, even when gromacs runs through fine... > ... for me - filesystem issues were causing this. In particular this > happened for me with NFS shares and on Lustre filesystems. My NFS > problems got solved with an upgrade of the NFS packages on our > machines (at least 1 1/2 yrs ago). > > Best > Martin > > > > ------------------------------ > > Message: 3 > Date: Mon, 22 Jun 2009 18:06:13 +0200 > From: Martin H?fling <[email protected]> > Subject: Re: [gmx-users] problems with some calculated trajectories > To: Discussion list for GROMACS users <[email protected]> > Message-ID: <[email protected]> > Content-Type: text/plain; charset=ISO-8859-1; format=flowed; delsp=yes > > > Am 22.06.2009 um 17:19 schrieb Martin Höfling: > > > > Hey Bernhard, here some thoughts of mine: > > In principle, vmd should be able to read the trajectories if pdb2gmx > tells you that the trajectory is ok. If this is not the case, you > can use gmx_rescue64 to extract the undamaged parts. > > > > ^ > ^^^^^^^^^^ > = gmxcheck :-) > > Best > Martin > > ------------------------------ > > Message: 4 > Date: Mon, 22 Jun 2009 13:29:10 -0400 > From: "Justin A. Lemkul" <[email protected]> > Subject: Re: [gmx-users] -bash: > /home_soft/soft_chem/soft_install/gromac/bin/GMXRC.bash: No such > file > or directory > To: Discussion list for GROMACS users <[email protected]> > Message-ID: <[email protected]> > Content-Type: text/plain; charset=GB2312 > > > > _______________________________________________ > gmx-users mailing list [email protected] > http://lists.gromacs.org/mailman/listinfo/gmx-users > Please search the archive at http://www.gromacs.org/search before posting! > Please don't post (un)subscribe requests to the list. Use the > www interface or send it to [email protected]. > Can't post? Read http://www.gromacs.org/mailing_lists/users.php > -- Tsjerk A. Wassenaar, Ph.D. Junior UD (post-doc) Biomolecular NMR, Bijvoet Center Utrecht University Padualaan 8 3584 CH Utrecht The Netherlands P: +31-30-2539931 F: +31-30-2537623 _______________________________________________ gmx-users mailing list [email protected] http://lists.gromacs.org/mailman/listinfo/gmx-users Please search the archive at http://www.gromacs.org/search before posting! Please don't post (un)subscribe requests to the list. Use the www interface or send it to [email protected]. Can't post? Read http://www.gromacs.org/mailing_lists/users.php
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}