The funny thing is, although we generally regard resolution as a primary
indicator of data quality the appearance of a density map at the classic
"1-sigma" contour has very little to do with resolution, and everything
to do with the B factor.
Seriously, try it. Take any structure you like, set all the B factors to
30 with PDBSET, calculate a map with SFALL or phenix.fmodel and have a
look at the density of tyrosine (Tyr) side chains. Even if you
calculate structure factors all the way out to 1.0 A the holes in the
Tyr rings look exactly the same: just barely starting to form. This is
because the structure factors from atoms with B=30 are essentially zero
out at 1.0 A, and adding zeroes does not change the map. You can adjust
the contour level, of course, and solvent content will have some effect
on where the "1-sigma" contour lies, but generally B=30 is the point
where Tyr side chains start to form their holes. Traditionally, this is
attributed to 1.8A resolution, but it is really at B=30. The point
where waters first start to poke out above the 1-sigma contour is at
B=60, despite being generally attributed to d=2.7A.
Now, of course, if you cut off this B=30 data at 3.5A then the Tyr side
chains become blobs, but that is equivalent to collecting data with the
detector way too far away and losing your high-resolution spots off the
edges. I have seen a few people do that, but not usually for a
published structure. Most people fight very hard for those faint,
barely-existing high-angle spots. But why do we do that if the map is
going to look the same anyway? The reason is because resolution and B
factors are linked.
Resolution is about separation vs width, and the width of the density
peak from any atom is set by its B factor. Yes, atoms have an intrinsic
width, but it is very quickly washed out by even modest B factors (B >
10). This is true for both x-ray and electron form factors. To a very
good approximation, the FWHM of C, N and O atoms is given by:
FWHM= sqrt(B*log(2))/pi+0.15
where "B" is the B factor assigned to the atom and the 0.15 fudge factor
accounts for its intrinsic width when B=0. Now that we know the peak
width, we can start to ask if two peaks are "resolved".
Start with the classical definition of "resolution" (call it after Airy,
Raleigh, Dawes, or whatever famous person you like), but essentially you
are asking the question: "how close can two peaks be before they merge
into one peak?". For Gaussian peaks this is 0.849*FWHM. Simple enough.
However, when you look at the density of two atoms this far apart you
will see the peak is highly oblong. Yes, the density has one maximum,
but there are clearly two atoms in there. It is also pretty obvious the
long axis of the peak is the line between the two atoms, and if you fit
two round atoms into this peak you recover the distance between them
quite accurately. Are they really not "resolved" if it is so clear
where they are?
In such cases you usually want to sharpen, as that will make the oblong
blob turn into two resolved peaks. Sharpening reduces the B factor and
therefore FWHM of every atom, making the "resolution" (0.849*FWHM) a
shorter distance. So, we have improved resolution with sharpening! Why
don't we always do this? Well, the reason is because of noise.
Sharpening up-weights the noise of high-order Fourier terms and
therefore degrades the overall signal-to-noise (SNR) of the map. This
is what I believe Colin would call reduced "contrast". Of course, since
we view maps with a threshold (aka contour) a map with SNR=5 will look
almost identical to a map with SNR=500. The "noise floor" is generally
well below the 1-sigma threshold, or even the 0-sigma threshold
(https://doi.org/10.1073/pnas.1302823110). As you turn up the
sharpening you will see blobs split apart and also see new peaks rising
above your map contouring threshold. Are these new peaks real? Or are
they noise? That is the difference between SNR=500 and SNR=5,
respectively. The tricky part of sharpening is knowing when you have
reached the point where you are introducing more noise than signal.
There are some good methods out there, but none of them are perfect.
What about filtering out the noise? An ideal noise suppression filter
has the same shape as the signal (I found that in Numerical Recipes),
and the shape of the signal from a macromolecule is a Gaussian in
reciprocal space (aka straight line on a Wilson plot). This is true, by
the way, for both a molecule packed into a crystal or free in solution.
So, the ideal noise-suppression filter is simply applying a B factor.
Only problem is: sharpening is generally done by applying a negative B
factor, so applying a Gaussian blur is equivalent to just not sharpening
as much. So, we are back to "optimal sharpening" again.
Why not use a filter that is non-Gaussian? We do this all the time!
Cutting off the data at a given resolution (d) is equivalent to blurring
the map with this function:
kernel_d(r) = 4/3*pi/d**3*sinc3(2*pi*r/d)
sinc3(x) = (x==0?1:3*(sin(x)/x-cos(x))/(x*x))
where kernel_d(r) is the normalized weight given to a point "r" Angstrom
away from the center of each blurring operation, and "sinc3" is the
Fourier synthesis of a solid sphere. That is, if you make an HKL file
with all F=1 and PHI=0 out to a resolution d, then effectively all hkls
beyond the resolution limit are zero. If you calculate a map with those
Fs, you will find the kernel_d(r) function at the origin. What that
means is: by applying a resolution cutoff, you are effectively
multiplying your data by this sphere of unit Fs, and since a
multiplication in reciprocal space is a convolution in real space, the
effect is convoluting (blurring) with kernel_d(x).
For comparison, if you apply a B factor, the real-space blurring kernel
is this:
kernel_B(r) = (4*pi/B)**1.5*exp(-4*pi**2/B*r*r)
If you graph these two kernels (format is for gnuplot) you will find
that they have the same FWHM whenever B=80*(d/3)**2. This "rule" is the
one I used for my resolution demonstration movie I made back in the late
20th century:
https://bl831.als.lbl.gov/~jamesh/movies/index.html#resolution
What I did then was set all atomic B factors to B = 80*(d/3)^2 and then
cut the resolution at "d". Seemed sensible at the time. I suppose I
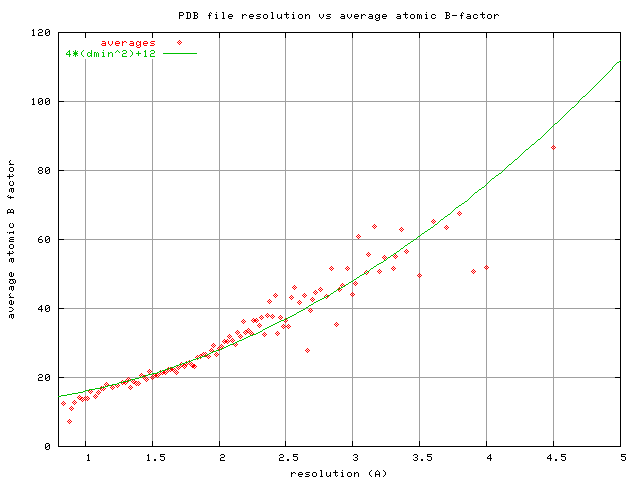
could have used the PDB-wide average atomic B factor reported for
structures with resolution "d", which roughly follows:
B = 4*d**2+12
https://bl831.als.lbl.gov/~jamesh/pickup/reso_vs_avgB.png
The reason I didn't use this formula for the movie is because I didn't
figure it out until about 10 years later. These two curves cross at
1.5A, but diverge significantly at poor resolution. So, which one is
right? It depends on how well you can measure really really faint
spots, and we've been getting better at that in recent decades.
So, what I'm trying to say here is that just because your data has CC1/2
or FSC dropping off to insignificance at 1.8 A doesn't mean you are
going to see holes in Tyr side chains. However, if you measure your
weak, high-res data really well (high multiplicity), you might be able
to sharpen your way to a much clearer map.
-James Holton
MAD Scientist
{kind=link}